Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Review Article - (2012) Volume 1, Issue 5
With the aging population and an increased incidence of neoplastic diseases, the study of the etiology of tumors and the mechanisms involved in the process of metastasis has increased significantly. Although there has been remarkable progress in this area and many proposed theories, several questions still remain. The factors associated with the emergence of the primary tumor and the mechanisms of metastasis are difficult to understand for many surgeons and clinicians because of the involvement of complex genetic, biochemical and molecular models. The tumor metastasis is responsible for approximately 90% of all cancer-related deaths. The cancer metastatic process occurs in a series of progressive steps, which called “metastatic cascade”. Its occurrence is a necessary break of the normal homeostatic mechanisms, leading to a rearrangement of the stromal tissue adjacent to the primary tumor. It is now well established that for this process, cancer cells need to acquire additional properties, which confer the capacity to invade the extracellular matrix, migrate, invade blood and lymph vessels, adhere, survive in target organs, grow and promote “organogenesis” in this new environment. The aim of this study, therefore, is to provide a didactic review and accessible information on ways and mechanisms involved in the genesis of the primary tumor and metastasis process, which may contribute to treatment of these tumors.
<Keywords: Cancer; Metastasis; Tumor
Genetic predisposition
The relationship between genetic predisposition and development of cancer is also well established. The familial cancer occurring due to mutations, which are genetically inherited, constitutes a small fraction (5%) of malignant tumors, but it creates high-risk for family members. Examples of the risks are retinoblastoma, breast carcinoma, Li Fraumeni syndrome (autosomal dominant disorder that predisposes to multiple forms of cancer, including breast, soft tissue sarcomas, brain tumors, osteosarcoma, leukemia, and adrenal carcinoma), and the syndrome of multiple endocrine neoplasms type 2 (MEN 2 – medullary thyroid carcinoma and pheochromocytoma, associated to mucosal neuromas or hyperplasia of the parathyroid). Recommendations for early diagnosis along with preventive examinations should be systematically applied and constitute the fundamental point to control the disease [1].
Genetic susceptibility
More than 110 different types of cancer have already been described, and the role of genetic susceptibility to carcinogenesis process is quite complex. It is known, however, that malignant tumor is a result of both genetic mutations and epigenetic changes, which interact, mainly, causing activation of proto-oncogenes and inactivation of suppressor genes of tumor [2].
Furthermore, because of the reduced visibility of cancer, all information necessary for the transformation of a normal cell in neoplastic must be attributed to modifications in its genome. It is now known that this genetic and phenotypic variability determines autonomous and unregulated cell growth, external signals that block and prevent the phenomena of apoptosis and favors the ability to invade adjacent tissues and organs, metastatic potential, and response or resistance to therapy. The number of genes implicated in tumor development is very high; in fact, although they act different initially, all the oncogenic agents have the same endpoint: transformation of proto-oncogenes into oncogenes and/or inhibition of anti-oncogenes [2,3].
Proto-oncogenes
Proto-oncogenes are groups of genes, which favor the conversion of a normal cell into a carcinogen cell when it is in mutation. Growth factors, membrane receptors, and DNA-binding proteins encode this process, and therefore, are related to growth, differentiation and proliferation of normal cells. Oncogenes are activated protooncogenes, and this process is triggered by means of the following genetic modifications [1,4].
Translocations and inversions – allow a proto-oncogene to be inserted near or added to a gene frequently transcripted, leading to its increased expression and/or aberrant production of proteins.
Deletions – have oncologic importance when they involve suppressor genes of oncogenic cell growth.
Amplifications – lead to the exacerbated expression of proteins structurally preserved.
Punctiform mutations – cause production of structural proteins and functionally aberrant.
Insertion of viral DNA – insert viral oncogenes in the human genome. The product of such oncogenes can stimulate or inhibit protooncogenes and anti-oncogenes.
The mechanisms of action of oncogenes have not been fully elucidated. Some oncogenes produce oncoproteins, such as, Bcl-2, IE84 (cytomegalovirus), SV40 T (SV40 virus), E6 (HPV), EBNA-5- (EBV), and HBx (hepatitis B virus), which strongly bind and inhibit proteins encoded by genes that suppress the growth or induce cell apoptosis, as the p53 and the Rb. As a result, they lead to the absence of suppression of the division or inhibition of cell death by apoptosis, and consequently to the cell “immortality” [2,4].
Other oncogenes act leading to overproduction of membrane receptors for growth factors such as c-erbB-2 (for a homologue of the epidermal growth factor) and RET. A third way is the autocrine production of growth factors, which is observed, for example, in the proliferation and activation of proto-oncogenes c-fos and c-sys by the product of the viral tax oncogene (HTLV-1) [5,6].
Other forms of promotion of tumor growth are the activation of proto-oncogenes that stimulate cell entry into mitosis (e.g., c-myc) and the production of proteins, which simulate the action of transducers of signal of the membrane receptors for growth factors (e.g., c-ras and c-abl) [5,6].
Anti-oncogenes
Anti-oncogenes are inhibitor genes of the normal and tumor cell proliferation. They operate in the following ways [2,7]:
• Interaction with the extracellular matrix – The anti-oncogene DCC produces a transmembrane protein that interacts with extracellular matrix components and is responsible for signaling growth inhibition by establishing contact between the cells lost in neoplasms.
• Regulation of transduction – The anti-oncogene NF-1 acts by inactivating protein of the proto-oncogene ras. This protein is a transducer whose function is to cause the core information of a cell; it is stimulated by growth factors bound to membrane receptors. In case of NF-1 inactivation by mutation or deletion, the transducer signal is not inhibited, generating a continuous stimulus to the cell into mitosis [2,7].
• Transcriptional regulation of DNA – The anti-oncogenes Rb and p53 are the prototype of this group. The gene Rb was the first anti-oncogene to be discovered during studies with the retinoblastoma. It acts by preventing the cell to get out of G0/G1 stages and enter into the S phase of cell cycle. When the cell undergoes mitogenic stimuli, the protein encoded by the Rb gene is inactivated, allowing the proliferative cycle progression; but, before the formation of the daughter-cells, it returns to its active form, thus, preventing the cycle from continuing indefinitely. When this gene is inactivated (e.g., by HPV and SV40), there is this block, and the cell reaches its “immortalization” [2,7].
P53 is one of the main genes responsible for the integrity of the genome. It is enabled by the emergence of altered DNA. Its activation produces a protein that stimulates the synthesis of others, which act by inhibiting cell replication by binding to the nuclear proliferation antigen (PCNA) and stimulating the DNA repair enzymes. If DNA repair is complete, the p53 is inactivated, and the cell returns to normal. If not satisfactory, the cell is prevented from replicating and is induced to apoptosis. Therefore, it plays a vital role in regulating, development, differentiation of the cell cycle, recombination of DNA, chromosome segregation and aging. It was named “guardian of the genome” and more than 50% of cancers are related to defects in the tumor suppressor gene p53 [7-10].
Age
Despite the genetic and environmental causes, age is the most important predictor of cancer development. About 80% of cancers arise in individuals aged over 55 years. The age-related inefficiencies in maintaining the integrity of the genome and the declining capacity of its repair is well studied and is nowadays critical to the emergence of the disease. This, therefore, means that the cancer that develops in old age represents a natural phenomenon of aging, which favors genomic instability and that cancer prevention should involve the complex task of reversing the modifications caused by aging [11,12].
Tumor microenvironment
Regardless of the route that enabled the development of the tumor, a dynamic microenvironment composed of tumor cells, stroma and extracellular matrix is established. The latter correspond to connective tissues of support, composed of several non-tumor cells such as fibroblasts, epithelial residing cells, myofibroblasts pericytes, vascular endothelial cells and linfovasculares, and infiltrating cells of immune system [3].
The recruitment of fibroblasts – the main component of the stroma – is responsible for the tumor initiation process. These cells, which are now called CAFS (cancer-associated fibroblasts), secrete factors that act on tumor cells of both paracrine and autocrine manner, which may favor the development of more aggressive tumor phenotypes. The CAFS secrete factors that promote the recruitment of immunosuppressive cells, creating a complex network which maintains the immunosuppressive environment. This scenario is responsible for immune escape of tumor cells and the consequent tumor progression [3].
Fibroblasts act primarily by depositing large amounts of extracellular matrix components, such as collagen type I and III and tenascin C. Moreover, the CAFS represent sources of metalloproteinase’s (MMPs) that promote tumor invasion by extracellular matrix degradation. Note the production of vascular endothelial growth factor and protein S100A4 that trigger the angiogenesis and plays a significant role in the process of metastases [13,14]. It has also been found that high levels of S100A4 expression correlate with the negative prognosis in various cancers [14].
Apoptosis
Apoptosis is the term used in contrast to necrosis, to describe the situation in which the cell is programmed to die in response to specific stimuli [10]. Three major biochemical changes are observed in apoptosis, including the activation of caspases, changes in the cell membrane with phagocytosis macrophages, and rupture of DNA and protein. One of the first changes in the observed phenomenon is the expression of phosphatidyl serine (PS) in the outer layers of the cell membrane. This allows for early recognition of dead cells resulting in phagocytosis by macrophages without release of pro-inflammatory cellular components. Then, the DNA fragmentation occurs with the subsequent internucleosomal cleavage by the endonucleases [15].
Another aspect is the activation of a specific group of proteins belonging to the family of cysteine proteases called caspases. Once activated, they degrade many vital cellular proteins; moreover, they disrupt the nuclear scaffold and cytoskeleton. The caspases are at the center of the apoptosis process, which they both initiate and execute. The two main described routes in its development are intrinsic (mitochondrial) and extrinsic (death receptor). At the end, both pathways may eventually lead to a common path named application pathway. Caspase 9 is the end part of the intrinsic pathway while caspase 8 is the extrinsic pathway. The intrinsic and extrinsic pathways converge to caspase 3, which enables nuclear apoptosis by disrupting the inhibitor of caspase-activated deoxyribonuclease [16].
The relationship between pro-apoptotic proteins and anti-apoptotic proteins, much more than their absolute values, play a fundamental role in the process of cell death and carcinogenesis. Therefore, overor under-expression of certain genes [Bcl-2, p53 Inhibitor of apoptosis proteins (IAPs)] have been implicated in carcinogenesis by reducing apoptosis in cancer cells. Apoptosis constitutes, therefore, of a highly selective process involved in physiological and pathological phenomena. Various ways in which malignant cells can acquire resistance to reduce the phenomena involved in apoptosis are described as follows: disrupted balance of pro-apoptotic and anti-apoptotic proteins reduced caspase function, and impaired death receptor signaling [17,18] (Figure 1).
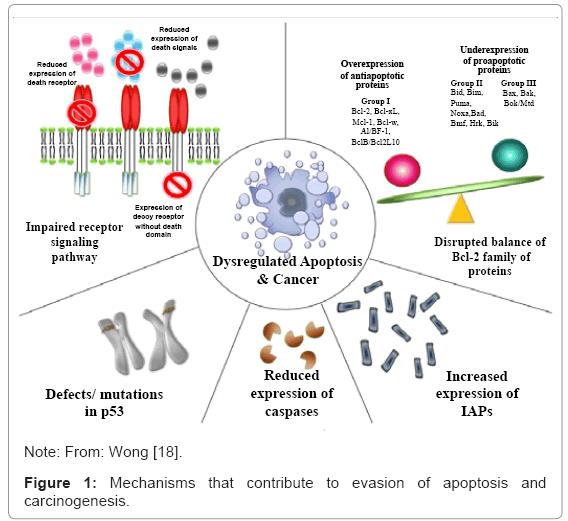
Figure 1: Mechanisms that contribute to evasion of apoptosis and carcinogenesis.
Steps in the metastatic process
The cancer metastasis is responsible for approximately 90% of all deaths related to this disease. Its occurrence is a necessary break of the normal homeostatic mechanisms, leading to a rearrangement of the stromal tissue adjacent to the primary tumor. A major trigger of this process is the hypoxia situation present in many tumor sites and corresponding to an oxygen pressure of the tissue below 5 for 10 mm Hg [13].
“Metastasis cascade” is the term used to describe a series of progressive steps that occurs during the metastatic process and includes: epithelial–mesenchymal transition (EMT) and breach of the basement membrane barrier; dissociation of tumor cells from the bulk tumor; invasion of the neighboring tissue; intravasation into preexisting and newly formed blood and lymph vessels; transport through vessels; extravasation from vessels; establishment of disseminated cells (which can stay dormant for a prolonged period of time) at a secondary anatomical site; and outgrowth of micrometastases and macrometastases/secondary tumors [19-23]. Moreover, another step, the creation of a “premetastatic niche” at the target site, before the first tumor cells arrive at this distant location, also is remembered. Each stage of the cascade triggers many physiological barriers to the spread of malignant cells and the tumor cells have to overcome all of them [19,20]. The different steps of “metastasis cascade” can be found in Figure 2.
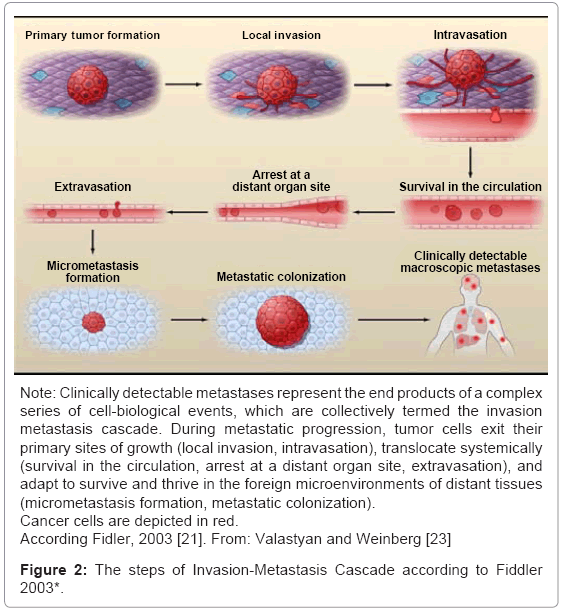
Figure 2: The steps of Invasion-Metastasis Cascade according to Fiddler 2003*.
Metastatic cascade
Cancer cell dissemination and epithelial–mesenchymal transition: Local invasiveness involves entry of cancer cells that have resided within a well-confined primary tumor into the surrounding tumor-associated stroma and thereafter into the adjacent normal tissue parenchyma [24]. Epithelial cells are the source of the majority of solid tumors and they are isolated from the stroma contact by a basement membrane (BM). They are also organized by lateral belts of cell– cell adhesion complexes. In order to invade the stroma, cancer cells must first breach the BM, a specialized ECM that plays vital roles in organizing epithelial tissues, in part by separating their epithelial and stromal compartments. During the progression from an “in situ” to an invasive tumor, cells are released from their neighbors and breach the basement membrane barrier. This phenomenon represents the first step in the metastasis cascade. The cancer cell loses its polarity and down regulation of epithelial proteins, mainly E-cadherin, but also occludin, claudins, cytokeratins or catenin proteins. Cadherins and catenins participate in cell–cell adhesion mechanism. Additionally, cells acquire a spindle-shaped morphology that allow cell migration and induce the production of mesenchymal proteins like N cadherin, vimentin, tenascin C, laminin â1 or collagen type VI á, as well as various proteinases [25,26] . The key signaling pathways and molecules inducing EMT, including Receptor Tyrosine, can directly modulate RTK signaling for instance by stimulating or repressing the activity of the epidermal growth factor receptor (EGFR) [27-29]. The induction of mesenchymal proteins during EMT also promotes invasive and metastatic processes: overexpression of N-cadherin, for example, induces cell migration, invasion and metastasis [30,31]. The snail and twist families of EMT mediators also inhibit apoptosis affecting both tumor growth and tumor spreading [32-35]. Recently has been shown that snail members mediate tumor immunosuppression and facilitating metastasis [36]. In addition, twist blocks cellular differentiation [37-39] and can interfere with oncogene induced senescence [40].
Invasion and cell migration: To invade tissues and vessels, cells must acquire the ability to migrate. The cell migration process starts with the extension of cell membrane protrusions, which is controlled by a continuous cycle of actin polymerization and depolymerization. After adhesion to the ECM via integrin- and FAK-containing complexes and actin–myosin 2-mediated cell contraction, release of adhesion at the trailing edge leads to cell locomotion. In this process, the cofilin pathway acts as the “steering wheel of the cell” by coordinating membrane protrusion [41]. Similarly, integrin signaling is critical for cell migration and invasion by modulating FAK/SRC signaling and the activity of RHO family GTPases [42].
Invasive tumor cells can migrate either as single cells or collectively in the form of files, clusters, or sheets. Collective invasion of tumor cells has been observed also in tumors with incomplete or no EMT. Single cell migration can occur either as a “mesenchymal” migration or in an “amoeboid” form, which is faster and requires no proteolytic ECM remodeling [43,44]. Many adhesion and signaling molecules, including integrins, CD44 and several Immunoglobulin-domain Cell Adhesion Molecules (IgCAMs), have been implicated in cell migration and tumor invasion [45,46]. EMT programs are orchestrated by a set of pleiotropically acting transcription factors, including Slug, Snail, Twist, ZEB1, and ZEB2, which organize entrance into a mesenchymal state by suppressing expression of epithelial markers and inducing expression of other markers associated with the mesenchymal state [47].
The invasion is considered a plastic process in which tumor cells can adapt to different conditions by switching their properties and requirements. Other concept is that the physical dimensions of ECM gaps and pores thereby determine the protease requirements, morphology and efficiency of cancer cell migration [48]. The main difference between metastatic and non-metastatic cells in that setting was not so much the migration capacity “per se”, but rather the directionality of migration. Metastatic cells become polarized towards blood vessels and migrated more directionally [49,50].
Anoikis: Once the cancer cells lose contact with the BM during invasion they face another barrier against metastasis: anoikis (cell death induced by inappropriate or loss of cell adhesion). It has already been shown that normal endothelial and epithelial cells actively trigger an apoptotic response once they lose their cell–cell and cell–matrix interactions or if the adhesive substrate is inadequate and this process ensure tissue homeostasis [51-53]. Thus, anoikis suppression is likely to be a prerequisite for tumor cells to successfully metastasize to distant sites [54,55]. Consistent with this, most cell lines established from human tumors contain populations of cells that survive when confronted with lack of adhesion to culture plates.
The main cell surface receptors to “sense” adhesion to the ECM and to provide a cell with information about its surroundings are the integrins [42]. Different integrin complexes bind to diverse ECM molecules and respond by triggering an intracellular signaling cascade via focal adhesion kinase (FAK) and SRC family kinases. Integrin activation protects cells against anoikis [52], similar to several kinases downstream of integrins, including SRC [51], focal adhesion Kinase FAK [56] and integrin linked kinase (ILK) [57,58]. Tumor cells often show an altered spectrum of integrin receptors [42,59] or have high levels of FAK [60], stimulating proliferation, survival and migration.
At same time, twist and snail are required for anoikis suppression in different cell systems [61,62]. Furthermore, the transcriptional corepressor C-terminal binding protein 1 (CTBP1) has been shown to repress both epithelial and pro-apoptotic genes at the same time [63], providing molecular insights into the connection between EMT and anoikis suppression. Besides suppressing anoikis by interfering with cell adhesion signaling, obstruction of the apoptotic machinery may also induce anoikis suppression and facilitate metastasis.
Angiogenesis: Tumor cell invasion alone is not sufficient to produce distant metastases; it requires also the transport of malignant cells through blood and/or lymph vessels. It is known that avascular tumors cannot grow beyond a size of 1 mm in diameter [64]. At this stage, passive diffusion of nutrients and oxygen becomes rate limiting for the tumor nodule, which is then forced to enter a state of so-called “tumor dormancy”. However, in most cases, tumor vascularization is achieved by sustained angiogenesis (sprouting of new vessels from existing ones), with a significant contribution of bone marrow-derived vascular and hematopoietic progenitor cells [65]. The growth of new vessels is strictly regulated by a delicate balance of angiogenic activators most prominently vascular endothelial growth factor A (VEGFA), fibroblast growth factors (FGFs), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF)) and angiogenic inhibitors (thrombospondin 1, angiostatin, endostatin and tumstatin) [66,67].
Under hypoxic conditions the cancer cells promote not only sustained angiogenesis but can also induce and select an invasive and metastatic phenotype and hypoxia-inducible factors (HIF1A, HIF2A) [13,68]. HIF1A regulates numerous target genes, including many that are involved in angiogenesis (VEGF), cell proliferation and glucose metabolism [69]. HIF1A can promote cell migration and invasion in different ways involving up regulation of the CXCR4 and up regulating lysyl oxidase (LOX) [70,71]. Furthermore, several EMT mediators, including twist, snail, ZEB1 and ZEB2, are induced by hypoxia and HIF1A in different cancer types [72-75]. Another aspect observed is a change in the normal vascular hierarchy of arterioles– capillaries– venules into a chaotic organization, leading to an abnormal blood flow that changes directions or even stops locally. In combination with increased leakiness of tumor vessels, this leads to a high interstitial (tissue) pressure in solid tumors and to an inefficient supply of nutrients and oxygen [76].
Tumor cells spread also via the lymphatic vasculature [77,78]. The presence of tumor cells in regional lymph nodes draining the primary tumor site can precede distant metastasis to visceral organs [77]. Most of the principles underlying tumor hemangiogenesis are conserved in lymphangiogenesis. For example, VEGF family members (VEGFC and VEGFD) induce lymphangiogenesis and lymph node metastasis via VEGF receptor 3 (VEGFR3) [79,80]. There are two possible explanations about why tumors attract lymph vessels in the first place, as, in contrast to blood vessels, they do not provide nutrients or oxygen and thus, do not seem to confer a direct advantage to the tumor.
The first one is that lymph vessels might lower the interstitial pressure in tumors. However, many intratumor lymph vessels seem to be non-functional [81]. Another one is that lymphangiogenesis represents merely a side effect; in that blood vessel endothelial cells release growth factors like FGF2 and PDGF, which not only stimulate tumor cell proliferation but also promote lymphangiogenesis [77].
Intravasation, transport through vessels and extravasation: The term intravasation describes invasive tumor cells entering the lumina of lymphatic or, mainly, blood vessels; process guided by macrophages and involving a paracrine signaling loop relying on the CSF1 receptor (expressed on macrophages) and EGFR (expressed on tumor cells) [82-84]. To facilitate the intravasation, a range of molecular changes can promote the ability of tumor cells to cross the pericyte and endothelial cell barriers, the microvessels walls’ components.
As soon as the cancer cells reach the lumina of blood vassels they disseminate widely through the venous and arterial circulation as CTCs (circulation tumor cells). However, while in the hematogeneous circulation, CTCs go through a series of stresses, such as sheer forces caused by the blood flow and the lack of cellular adhesion. As a result, a large number of tumor cells undergo anoikis, eliminating disseminated tumor cells and hampering metastasis.
It is not known how long the CTCs that could survive those stresses can circulate in the vasculature. Some studies suggest that, due to the relative large diameter of cancer cells in comparison with the luminal diameter of capillaries, CTCs are trapped in the first or second capillary bed they find [19]. In this way, many tumor cells just spend a short period in the hematogeneous circulation, escaping from the anoikis inducing mechanisms.
Once located in the microvassels of distant sites, the circulating tumor cells initiate their extravasation, crossing from the vessel lumina into the tissue parenchyma. During this process, integrins ans selectins promote the interaction of tumor cells with platelets, leukocytes and endothelial cells, allowing CTCs penetration trough the layers of pericytes and endothelial cells that separate vessel lumina from the stromal microenvironment [85,86].
Micrometastasis formation: When tumor cells extravagate, they encounter a foreign microenvironment formed by stromal cells, ECM constituents, available growth factors and cytokines that usually differs from that one of the primary tumor. In order to survive and form micro metastasis, tumor cells use effective mechanisms to modify the metastatic site properties. According to the “premetastatic niche” model, before the arrival of disseminated tumor cells, the primary tumor releases systemic signals (perhaps lysyl oxidase) that promote a range of changes and convert distant sites into more hospital environments for the survival of those tumor cells and the formation of micrometastases [87]. Simultaneously, metastatic cells can adapt themselves to the new environment by using cell-autonomous processes. One example of such a mechanism involves activation of Src tyrosine kinase signaling [88].
Metastatic colonization: It seems that, in the process of metastatic colonization, the majority of disseminated tumor cells suffers either slow attrition over periods of weeks and months or persists as microcolonies in a state of apparent long-term dormancy. The disseminated cancer cells may be quiescent, with their proliferation at metastatic sites greatly impaired due to incompatibilities with the foreign microenvironments that surround them [19].
Moreover, the ability of disseminated tumor cells to escape dormancy and to begin active proliferation may depend on cellnon autonomous mechanisms that are needed to convert foreign microenvironments into more hospitable niches. The outgrowth of indolent disseminated cancer cells may depend on the activation and mobilization into the circulation of bone marrow-derived cells and the subsequent recruitment of these cells to a metastatic site. In some cases, these processes may be stimulated by systemic signals released by carcinoma cells, such as osteopontin (OPN) or SDF-1[89,90].
On the other hand, the occult micro metastases may proliferate continuously; however, a net increase in their overall number may not occur due to the effects of a high apoptotic rate. The failure of the occult micro metastasis to initiate neoangiogenesis has been proposed as explanation for this high attrition rate [19].
The ‘‘seed-and-soil’’ hypothesis [91] of metastatic outgrowth articulated more than 120 years ago is still current. More recently, a number of genes whose expression facilitates the metastatic colonization of breast cancer cells specifically to both lung [92] and brain [93], have been identified. These genes seem to dictate organspecific metastatic tropism due to their ability to compensate for and overcome incompatibilities between the intrinsic growth programs of the disseminated cancer cells and the demands imposed by the particular foreign tissue microenvironment around them [23].
Therefore, the final step of the invasion-metastasis cascade imply that the distinct adaptive programs governing metastatic colonization may number in the dozens, with each determined by both (1) the identity of the organ site at which metastatic colonization occurs and (2) the tissue of origin of the disseminating primary tumor cells; in other words cancer cells colonizing the lungs utilize different genetic and/or epigenetic programs than do the same breast carcinoma cells colonizing the bone, brain, or liver [23].
Other relevant aspect to metastatic colonization, the ‘‘tumorinitiating cells’’ TIC hypothesis asserts that one or more self-renewing TICs must disseminate during the course of disease progression in order for macroscopic metastases to develop; conversely, the limited self-renewal capacity of disseminated non-TICs may preclude them from spawning macroscopic metastases. One class of molecules that promote entrance into the TICstate is EMT-promoting transcription factors, such as Snail,Twist, and ZEB1. This interaction between a molecular pathway that promotes both invasiveness and self-renewal is noteworthy, as these transcription factors appear to concomitantly facilitate physical dissemination of cancer cells and, following dissemination, the proliferation of these cells at distant organ sites [23].
Lastly, the accumulation of genetic and/or epigenetic alterations, as well as the co-option of nonneoplastic stromal cells, cancer cells are capable of completing an intricate, multistep, cell-biological process that culminates in the formation of macroscopic, life-threatening growths at distant organ sites [23] (Figure 3).

Figure 3: Inefficiency of the” Invasion-Metastasis Cascade”.
Others relevant phenomena in cancer metastasis
Epigenetic changes: Among the main molecular phenomena involved in the metastatic process are epigenetic changes, which are able to explain the alterations suffered by many genes involved in metastasis, as the genes encoding laminins, heparan sulfates, proteases, inhibitors of angiogenesis, among others. The term epigenetics refers to the inheritance of changes in the gene activity that is independent of the sequence of DNA [94]. Two main mechanisms are involved in epigenetic gene regulation in the development and genesis of tumors: the hypermethylation of DNA, and the changes undergone by histonas [95]. Such modifications are indicative of a cancerous process, in which the expression of tumor phenotypes occurs because of loss of fine control of the epigenetic mechanisms [96].
The epigenetic methylation corresponds to a stable change of the genetic expression through changes in genomic DNA by the addition of a methyl group to cytosine residues or adenine. A gene can then be inactivated, i.e., having its transcription inhibited by the hypermethylation of its promoter region. Thus, the genesis and tumor progression are linked to the epigenetic inactivation of the promoter regions of suppressor genes of tumors [97]. In addition, aberrant epigenetic alterations are used as biomarkers for diagnosis, staging, and response to tumor treatment.
The early research studies in this area have observed the presence of methylation in cytosine positioned immediately after guanine residues (known as CpG). In somatic cells of an adult, 60–90% of the CpG are methylated, with the remainder, unmethylated, known as “CpG islands”. The core of the genesis and tumor progression is exactly the suppression by hypermethylation of these islands “CpG” [98]. To evaluate the methylation of “CpG islands”, several techniques are used, including the specific-methylation PCR (MSP).
Surprisingly, DNA methylation occurs in a complex context of chromatin and is influenced by the changes that occur with the histones. It is known that histones are dynamic regulators of genetic activity, which are subject to a series of post-transcriptional chemical modifications, such as acetylation, methylation, phosphorylation and ubiquilation [94]. In general, some modifications such as acetylation of histones are responsible for promoting its transcriptional activity, while others, such as methylation of lysine 9 of histone H3, cause condensation and inactivation of chromatin [99]. According to the hypothesis of the “histone code”, the expression profile of a given region of chromatin depends on the combination of histone modifications [100].
Changes involving microRNAs: In addition to DNA hypermethylation and modifications suffered by histone, the epigenetic changes involving are extremely important in tumor development and metastasis. MicroRNAs (or miRs) are non-coding RNA molecules comprising of 19–25 nucleotides, capable of regulating the genetic expression [101]. The action of the miRs is the inhibition, and it occurs when partially complementary sequences are present in a 3' nontranscribed region of a mRNA target or when the miR and the target mRNA bind, causing the degradation of the mRNA and consequent inhibition of the translation. The miRs have an important role during mammals’ development in the processes of proliferation, apoptosis and differentiation [101]. Nowadays, there is new evidence that there is an unregulated expression of miRs by epigenetic mechanisms in human cancers. The pathogenesis of tumor occurs either by overexpression of miRs, causing hipoexpression of the regulatory genes of tumors, or by the miRs hipoexpression, causing overexpression of oncogeneses [102].
The chemokines: In the intricate metastatic process, the role of chemokines is very important; pro-inflammatory cytokines promote the immediate chemotaxis of leukocytes to the inflammatory site. They are induced by inflammatory cytokines, growth factors, and pathogenic stimuli signaling through chemokine receptors (7-transmembrane coupled to G-protein) [103]. They are classified into four groups (C, CC, CXC, and CX3C) according to the position of the two residues N-terminal of cysteine, and family to which their binder cognates belong [104]. Such molecules can be expressed by different cell types and control many physiological and pathological processes, such as, metastasis of malignant cells to distant organs. Since cytokines and chemokines are responsible for the inflammatory process in this tumor genesis, they contribute to the promotion of angiogenesis, tumor growth, invasion and metastasis [105].
Organotropism: Nowadays, it is consensual that the probability of a tumor metastasis is a non-random event, which depends on the interaction with tumor factors that facilitate their homeostatic proliferation and survival. There are numerous theories that seek to explain the organotropism present in the metastatic process, which is partly explained by the pattern of blood flow. In the late nineteenth century, Paget developed the hypothesis of “seed and soil” [91]. Noting the susceptibility of tumors to migrate to specific sites, he proposed that certain tumor cells (the seeds) select bodies, whose microenvironment is propitious to their growth (the soil). The theory of “selection of the organ” considers that tumor cells migrate in proportion to the primary site for the entire circulation and/or lymph nodes. In this case, what determines the establishment of metastases in target organs is the presence of appropriate growth factors in such locations. The theory of “adhesion” suggests that adhesion molecules expressed in tissue-specific endothelial surface of target organs are responsible for anchoring migratory tumor cells, and create a pre-tumor niche that had become a secondary tumor. The theory of “chemoattraction” proposes that malignant cells expressing functional chemokine receptors can respond to organ-specific molecules and migrate directionally by gradients of chemokines and initiate site-specific metastases in target organs [106].
Molecular and cellular mechanisms of metastasis
There are countless efforts to elucidate the molecular and cellular mechanisms governing the metastatic process. A current concept suggests the existence of tumor stem cells, or CSC’s, which are undifferentiated cells, responsible for the initiation, growth and ability to metastization and recurrence of a tumor. They can differentiate into several cell types of primary tumor and have the ability to self-renew and maintain stem cells and tumor cells mature indefinitely; they resemble the normal stem cells. Such cells are likely to result from several mutations on normal stem cells, tissue, or bone marrow. Moreover, its origin can be associated with the process of trans-differentiation of somatic cell or to the process of genetic transfer [107].
Since the molecular and cellular mechanisms of metastasis remain clouded by many questions, there are several models that attempt to explain them:
(A) “Cell-of-Origin Model”. The normal differentiation programs of the cells of origin from which certain primary tumors are derived may already dictate the altered activity of various metastasis virulence genes (depicted in green). Upon subsequent oncogenic transformation and systemic dissemination, these cells may therefore be capable of completing the process of metastatic colonization [108] (Figure 4A).
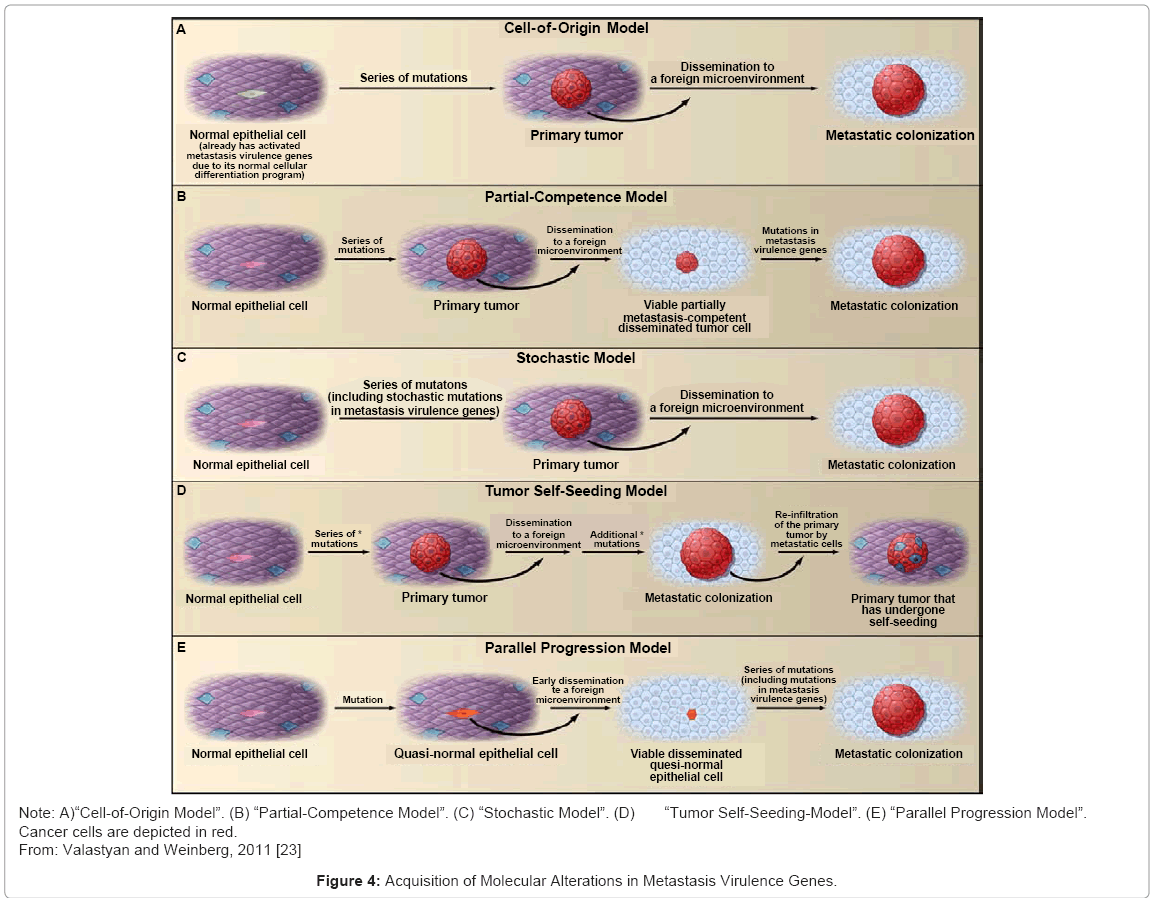
Figure 4: Acquisition of Molecular Alterations in Metastasis Virulence Genes.
(B) “Partial-Competence Model”. Cells that are only partially metastasis competent (that is tumor cells that have acquired a series of mutations that confer the capacity to disseminate systemically but are initially unable to colonize foreign microenvironments) may arrive at distant organs, where they then undergo further genetic and/ or epigenetic evolution within these foreign microenvironments to achieve full metastatic competence. Such molecular evolution would likely include alterations in metastasis virulence genes [108] (Figure 4B).
(C) “Stochastic Model”. Purely by chance, mutations in metastasis virulence genes may accumulate stochastically as ‘‘passenger mutations’’ within tumor cell clones that bear unrelated ‘‘driver mutations’’ that serve to fuel the clonal expansion of these cells within primary tumors [109] (Figure 4C).
(D) “Tumor Self-Seeding-Model”. The phenomenon of tumor self-seeding indicates that already metastasized cells are capable of re-infiltrating the primary tumor from which they originated. Hence, carcinoma cells present in metastases (which have come to acquire molecular alterations in metastasis virulence genes via either of the models proposed below, as indicated by the asterisk) may become increasingly represented within their primary tumor of origin (reinfiltrating cells are depicted in blue) [110] (Figure 4D).
(E) The “Parallel Progression Model”. Asserts that quasi-normal epithelial cells (depicted in orange) disseminate very early from preneoplastic lesions. Subsequently, these cells undergo molecular evolution at future sites of metastasis formation. Notably, such sites represent locations where mutations in metastasis virulence genes are now selectively advantageous [110,111] (Figure 4E).
Lymph node metastasis
Patients with cancer are at a high risk of local disease recurrence and distant metastasis developing after the local surgical resection process, often simultaneously with lymphadenectomy, indicating when there is the possibility of lymph node metastasis. Besides being a prognostic marker, the presence of metastatic cells in lymph nodes, is related to the development of distant metastases [112,113].
In general, cancer progression is consistent with Hellman’s spectrum theory in that development of nodal and systemic metastasis from a localized cancer growth is a progressive process. Nevertheless, in about 20 % of the time, the cancer cells may spread directly through the blood vascular system to the distant sites. The process of cancer cells spread to the regional sentinel lymph node first, then beyond to the systemic sites, has been well established in melanoma and breast cancer [113,114].
However, little is known about the molecular mechanism of lymph node metastasis, generating doubts regarding the possibility of tumor cells accumulation in the lymph nodes, delaying the spread of tumor, the possibility of lymph nodes disseminating tumor cells, amplifying them and throwing them to the bloodstream, and the possibility of tumor cells only being evidence of the cell traffic by the lymph nodes [114].
Therefore, many theories have been postulated to explain the intricate mechanism of tumor appearance and metastatic process, but none of them completely explain all biological and clinical observations. Consequently, metastasis still remains an open issue with only few metastasis-inducing proteins experimentally validated so far.
Carlos Augusto Gomes, Cleber Soares Junior, Juliana Almeida do Valle, Carolina Teixeira de Assis Lopes, Andressa Barra, Camila Couto Gomes, Thiago Henrique Ladeira do Carmo and Felipe Couto Gomes, have no conflicts of interest or financial ties to disclose.