Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2013) Volume 1, Issue 3
Human C1-esterase inhibitor (C1-INH) is a unique anti-inflammatory multifunctional plasma protein best known for its key role in regulation of the classical complement pathway, contact activation system and intrinsic pathway of coagulation. By sequence homology and mechanism of protease inhibition it belongs to the serine proteinase inhibitor (serpin) superfamily. However, in addition to its inhibitory capacities for several proteases, it also exhibits a broad spectrum of non-inhibitory biological activities. C1-INH plays a key role in the regulation of vascular permeability, best demonstrated in Hereditary Angioedema (HAE) which is triggered by the deficiency of functional C1-INH in plasma? Since 1963, when the link between HAE and C1-INH was first identified, considerable progress has been made in the investigation of C1-INH structure and biological activities, understanding its therapeutic potential, and in the research and development of C1-INH-based therapies for the treatment of HAE and several other clinical conditions. However, augmentation therapy with C1-INH concentrates for patients with HAE is currently the only approved therapeutic application of C1-INH. This manuscript provides an overview of the structure and functions of human C1-INH, its role in HAE, summarizes published data available for recently approved C1-INH therapeutic products, and considers possible use of C1-INH for other applications.
Keywords: C1-esterase inhibitor; C1-inhibitor deficiency; Hereditary angioedema
BK: Bradykinin; B2R: B2-receptor; C1-INH: C1- esterase inhibitor; FXIIa: activated factor XII; GAG: Glycosaminoglycan; HAE: Hereditary Angioedema; HMWK: High Molecular Weight Kininogen; IV: Intravenous; MASP-1 and MASP-2: Mannan-Binding Lectin-Associated Serine Proteases; pd: Plasma-Derived; RCL: Reactive Center Loop; Rhc1-INH: Recombinant Human C1-INH; SC: Subcutaneou
Human C1-INH is an important anti-inflammatory plasma protein with a wide range of inhibitory and non-inhibitory biological activities. By sequence homology, structure of its C-terminal domain, and mechanism of protease inhibition, it belongs to the serpin superfamily, the largest class of plasma protease inhibitors, which also includes antithrombin, α1-proteinase inhibitor, plasminogen activator inhibitor, and many other structurally similar proteins that regulate diverse physiological systems [1,2]. Best known for regulating the complement cascade system, C1-INH also plays a key role in the regulation of the contact (kallikrein-kinin) amplification cascade, and participates in the regulation of the coagulation and fibrinolytic systems [3-5]. Since 1963, when Donaldson and Evans linked the symptoms of hereditary angioedema (HAE, called hereditary angioneurotic edema at that time) to the absence of serum C1-INH, it has been the subject of multidisciplinary research [6,7]. Over the past fifty years, our knowledge about C1-INH structure and its biological functions, the cause(s) of its deficiency, and its role in HAE and other conditions has been significantly advanced. The majority of research still focuses on the mechanisms and efficient treatments of C1-INH-dependent types of HAE; however, the physiological and pharmacological activities of C1-INH are much broader. During the last five years, several C1-INH products have been licensed in the US and EU for the treatment C1-INH-deficient patients with HAE that dramatically improved the therapeutic options in the battle with this rare disease. Due to C1-INH involvement in a variety of physiological processes, its therapeutic potential for the treatment of other conditions has also been recognized, yet remains unexplored. This review considers the biochemistry of C1-INH, its biological activities, and recent advances in its therapeutic development.
Abundance of C1-INH
Human C1-INH is a positive acute-phase plasma glycoprotein with normal concentration in healthy subjects ~ 240 μg/mL (~3 μM), but its level may increase by 2-2.5 times during inflammation [8,9]. Some characteristics of human C1-INH are summarized in table 1. Low plasma content of C1-INH or its dysfunction result in the activation of both complement and contact plasma cascades, and may affect other systems as well (vide infra) [10-12]. Decrease in C1-INH plasma content to levels lower than 55 μg/mL (~25% of normal) was shown to induce spontaneous activation of C1 [13].
| Characteristics | Description |
|---|---|
| Synonyms | C1-esterase inhibitor, C1s-inhibitor, C1-inactivator, α2-neuramino-glycoprotein |
| Common abbreviations | C1-INH, C1INH, C1-Inh |
| Classification | Serine proteinase inhibitor (serpin) |
| Substrates | C1s, C1r, Plasmin, Kallikrein, aFXIIa, bFXIIa, FXIa, MASP-1 and MASP-2 |
| Molecular weight | ~74 kDa by mass spectrometric analysis; ~76 kDa by neutron scattering; ~105 kDa by SDS-PAGE |
| Polypeptide | Single polypeptide chain of 478 amino acid residues |
| Domains | Two-domain structure: C-terminal serpin domain (362 amino acid residues), and N-terminal domain (~113) |
| Glycosylation | N- and O- attached carbohydrates (26% w/w) |
| Heterogeneity | Highly heterogeneous protein |
| Half-life in circulation | ~28 h (in healthy volunteers)a, 67.7± 4.9 h (in HAE patients)b |
| Concentration in blood | ~240 mg/L (in healthy volunteers), may increase 2-2.5 times in response to inflammation |
| Major biological activities | Inhibitory anti-serine proteinase activity Multiple non-inhibitory activities |
| Physiologically important mutants | 237 different single mutations related to the deficiency or dysfunction of C1-INH in HAE patientsc |
| Known cofactors | Glycosaminoglycans |
Table 1: Characteristics of Human C1-INH.
Biosynthesis and mutations
C1-INH is synthesized and secreted primarily by hepatocytes, but is also produced by monocytes, fibroblasts, macrophages, microglial cells, endothelial cells, and some other cells [14-18]. Biosynthesis of C1-INH can be stimulated by cytokines, particularly by interferon-γ [19,20].
Human C1-INH is encoded by the SERPING1 gene (17 kb) on chromosome 11 (11q12→q13.1) comprised of eight exons and seven introns [21-23]. A vast variety of large and small mutations identified in C1-INH for patients with HAE results from three types of alterations in the gene: (i) deletions or duplications due to high content of Alu repeat sequences in the introns that causes ~20% of genetic defects in HAE [23,24]; (ii) mutations in the codon for Arg444 of the reactive center loop (RCL) which results in dysfunctional C1-INH [25]; and (iii) a large number of single codon mutations over the length of the gene [12,26-28]. All this characterizes C1-INH deficiency as an extremely heterogeneous disease. It is of interest that some mutations in the SERPING1 gene are associated with the development of age-related macular degeneration [29,30].
Structural characterization
Human C1-INH is a single-chain polypeptide (478 amino acid residues) which forms two domains: (a) C-terminal domain (365 amino acids), which is a typical serpin; and (b) N-terminal domain (113 amino acids), the role of which is not clearly elucidated, but which seems to be important for protein integrity and stabilization of the serpin domain [31] and for interactions with lipopolysaccharides [32].
C1-INH is a heavily glycosylated protein with more than 26% (w/w) of carbohydrates. Whereas the calculated molecular weight of the polypeptide part is 52,869 Da [21], the molecular weight of the whole protein is ~76 kDa (by neutron scattering [33]). Using mass spectrometric analysis we detected it as an ~74 kDa protein (the author’s data); however, it is often referred to as an ~105 kDa glycoprotein as observed by electrophoretic mobility on SDS-PAGE [33,34]. Carbohydrates are very unevenly distributed over the C1-INH molecule: (1) three N-attached glycans belong to the serpin domain, attached to asparagine residues Asn216, Asn231, and Asn330, which is similar to the glycosylation of archetypal serpin α1-proteinase inhibitor; and (2) three N- and at least seven O-linked carbohydrates have been determined for N-terminal domain [35]. The protein integrity is maintained by connection of the domains with two disulfide bridges formed by Cys101 and Cys108 of the N-terminal domain and Cys406 and Cys183 of C-terminal domain [31]. Although the functional role of the exceptionally long and heavily glycosylated N-terminal domain is still unclear, it may be essential for the protein’s conformational stability, recognition, affinity to endotoxins and selectins, and clearance. The intrinsic heterogeneity of the carbohydrate moiety greatly contributes to the heterogeneity of the whole C1-INH. This may be one of the reasons why obtaining a crystal structure of native glycosylated C1-INH remains elusive.
To date, the only published crystal structure that is available for C1-INH has been performed by Beinrohr and co-workers for the non-glycosylated serpin domain of recombinant C1-INH, which crystallized in its latent form [36]. The crystal structure of the serpin domain features nine α-helices, three β-sheets, and a mobile RCL exposed for interaction with target serine proteases, features typical for serpins (Figure 1). The alignment of the C1-INH serpin domain structure with those of antithrombin III and α1-proteinase inhibitor illustrates the overall remarkable structural similarity between serpins [37]. The major difference revealed by Beinrohr et al. for this C1-INH serpin structure is that the C-terminal P’ part of the RCL shows as the new seventh strand of thus-extended β-sheet A, while in other known latent serpin structures it is just a flexible loop [36].
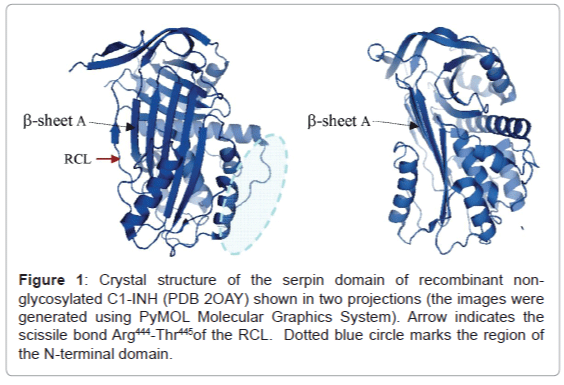
Figure 1: Crystal structure of the serpin domain of recombinant nonglycosylated C1-INH (PDB 2OAY) shown in two projections (the images were generated using PyMOL Molecular Graphics System). Arrow indicates the scissile bond Arg444-Thr445 of the RCL. Dotted blue circle marks the region of the N-terminal domain.
Similar to α1-proteinase inhibitor, C1-INH serpin domain is intrinsically folded into a metastable structure, which is not the most thermodynamically stable form, but is essential for inhibitory function. Like other inhibitory serpins, C1-INH neutralizes target proteases in a suicide fashion [7,38,39]. By attacking the RCL of the serpin, protease cleaves the scissile bond Arg444-Thr445, consistent with protease substrate specificity. Formation of the covalent complex between the protease and C1-INH is followed by a drastic conformational perturbation that results in the translocation of the protease over the C1-INH to the opposite side of the molecule, thus inserting the cleaved RCL into β-sheet A of the inhibitor.
Inhibition of proteases and other activities
C1-INH exhibits its inhibitory potential towards several proteases, and thus, plays a unique role in the down-regulation of four major plasmatic cascade systems. It is the only regulator of the early proteases of the classical complement pathway activation (C1s and C1r) [34,40,41]. By the inactivation of mannan-binding lectin-associated serine protease-2 (MASP-2), it also participates in the regulation of lectin pathway activation (in addition to α2-macroglobulin) [42,43]. Being the primary regulator of the contact system, C1-INH inactivates the activated plasma kallikrein and FXIIa; the inhibition of kallikrein by C1-INH proceeds with a much higher rate than that of α2-macroglobulin [44-46]. C1-INH also participates in the regulation of the fibrinolytic pathway by the inactivation of plasmin and tissue plasminogen activator [47], as well as it seems to be involved in the inhibition of thrombin and FXIa of the intrinsic coagulation pathway [48,49].
In addition to its inhibitory potential, C1-INH also possesses a broad spectrum of non-inhibitory activities, including interactions with endogenous proteins, polyanions (glycosaminoglycans), various types of cells and infectious agents [7,50-52]. Figure 2 summarizes C1-INH major biological activities known so far.
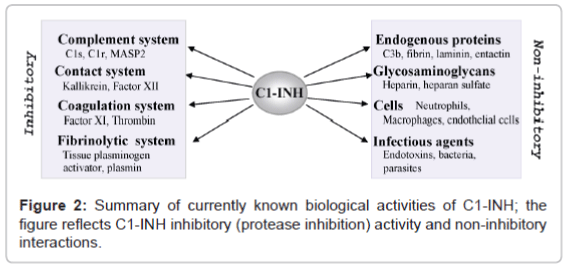
Figure 2: Summary of currently known biological activities of C1-INH; the figure reflects C1-INH inhibitory (protease inhibition) activity and non-inhibitory interactions.
Potentiation by glycosaminoglycans
It is known that inhibitory activities of some serpins, including ATIII and C1-INH, are modulated by glycosaminoglycans (GAGs), particularly by heparin [53-55]. The potentiation of C1-INH by GAGs significantly enhances its inhibitory activity toward C1s and factor XIa (FXIa), but barely to C1r [56-59]. Over the last two decades, C1-INH interactions with various physiological GAGs (heparin, heparan sulfate, chondroitin sulfate, dermatan sulfate) and non-physiological GAG derivatives (dextran sulfate, oversulfated chondroitin sulfate) have been under intensive investigation to define the influence of C1-INH potentiation on the complement, and other plasmatic, cascades [50,60-63].
Notably, no GAG-modulated influence on the inhibition of kallikrein and FXII by C1-INH was observed [62]. The available data suggest that C1-INH potentiation by GAGs is selective toward target proteases, and thus may selectively enhance the inactivation of the classical complement pathway and the intrinsic pathway of coagulation (via C1s and FXIa, respectively) without significant impact on fibrinolytic activity of activated factor XII or kallikrein.
In 2007 and 2008, multiple adverse events were reported in the US and elsewhere in the world caused by transfusions of heparin that was contaminated by oversulfated chondroitin sulfate [64,65]. Further investigation revealed that while heparin can inhibit the complement system via potentiation of C1-INH, the oversulfated chondroitin sulfate is much more potent inhibitor of complement than heparin [37,66].
Despite the structural similarity between serpins, the proposed mechanism of the C1-INH potentiation by GAGs (a “sandwich” mechanism), as well as a putative heparin binding site on C1-INH surface, differ from those of antithrombin III [36], whereas the archetypal serpin α1-proteinase inhibitor appears not to be a subject of GAG potentiation at all. According to the model proposed by Beinrohr et al. [36], by binding to C1-INH and neutralizing its positively charged surface patches the GAGs facilitate its interaction with C1s. To date, antithrombin remains the only serpin the enhancement of which by heparin (up to 4000-fold enhancement) has been further developed for use in current clinical practice. It is therefore feasible that currently available or emerging C1-INH-based therapies can also be enhanced by GAGs.
Due to the multiple biological activities of C1-INH and its essential role in many physiological processes, the pharmaceutical potential of C1-INH is attractive and promising, yet not easy to explore since C1-INH is involved in several interrelated plasmatic cascade systems. Recent years brought a dramatic breakthrough in the therapeutic development of C1-INH. Several C1-INH products became available for replacement (augmentation) therapy in C1-INH-deficient patients with HAE, leading to a marked reduction in morbidity and mortality.
C1-INH and HAE
The importance of functionally active C1-INH is clearly exemplified by its deficiency, considered to be the cause of HAE, a rare autosomal dominant disease (with an estimated frequency of 1:50 000) that is clinically manifested by recurrent acute attacks. The release of bradykinin (BK) caused by low content or dysfunction of C1-INH results in increased vascular permeability and edema in subcutaneous or submucosal tissues at various body sites (face, extremities, genitals, abdomen, oropharynx or larynx) [67-69]. These acute attacks cause pain and disability during subcutaneous and intestinal episodes, and are potentially life- threatening when edema strikes upper airways [70,71].
Although the descriptions of HAE appeared in the 19th century, with the first article published on the subject in 1888, the link between C1-INH and HAE was identified only in 1963 (reviews [72,73]). Abnormalities in C1-INH plasma content or in its functional activity (often referred to as a deficiency of functional C1-INH) result from various large and small mutations in the C1-INH gene (vide supra). There are generally two types of hereditary C1-INH deficiency. The more prevalent type I HAE (approximately 85% of HAE cases) is characterized by low content (below 35% of normal) and low inhibitory activity of C1-INH in the circulation. Type II HAE (approximately 15% of HAE cases) is associated with normal or elevated antigenic levels of C1-INH of low functional activity [68,74]. Recently, HAE with normal C1-INH (also known as type III HAE) has been described in two subcategories: (1) HAE due to mutation in the factor XII gene and, as a result, increased activity of factor XII leading to a high generation of BK, and (2) HAE of unknown genetic cause [75-77]. Contrary to hereditary angioedema, acquired angioedema (AAE) is associated with lymphoproliferative disease and/or C1-INH reactive autoantibodies that neutralize its inhibitory activity [78]. (For more details on HAE see comprehensive reviews [73,79-81]). Whereas activation of the contact system leads to the onset of BK-mediated acute attacks directly, the imbalance of C1-INH level greatly depends upon its consumption by other plasma cascades, also regulated by C1-INH, that may influence the activation of the contact pathway leading to edema, as well [69,82,83]. Figure 3 depicts major plasmatic cascades regulated by C1-INH to illustrate that C1-INH interactions with several target proteases of these plasmatic cascades suggest rapid depletion of functionally active C1-INH, leading to the development of angioedema, as well as resulting in possible overlap of highly diverse outcomes from the activation of the complement and other systems [82,83]. Whereas BK is the major mediator of vascular permeability and edema, a large body of evidence supports complex interrelationships between contact, complement, coagulation and fibrinolytic systems. During HAE attacks in C1-INH-deficient patients, low C2 and C4 levels indicate that the complement is activated, while increased levels of thrombin and generation of plasmin indicate that the coagulation and fibrinolytic systems are involved [84-87].
During the last five years treatment options for HAE patients in the US have been dramatically revolutionized. Until recently these patients were treated with transfusions of fresh-frozen plasma (which contains endogenous C1-INH), despite potential risk of blood-borne pathogens, and with antifiblinolytics and attenuated androgens, despite frequent adverse effects due to residual hormonal activity of androgens [88-90]. Currently, there are several therapies that are licensed specifically for treatment of HAE acute attacks or prophylaxis (Table 2) [91-93]. Owing to the genetic origin of HAE, these treatments do not cure the disease per se, but are rather intended to relieve symptoms and pain associated with acute attacks and to prevent the frequency and severity of HAE episodes. Considering HAE as a BK-mediated swelling disorder caused by C1-INH deficiency, current therapeutic strategies target three key steps in the contact (kallikrein-kinin) pathway leading from reduced levels of functional C1-INH to BK-induced edematous conditions, i.e.: (a) C1-INH replacement to restore plasma levels of functional C1-INH; (b) Inhibition of plasma kallikrein to prevent BK formation; and (c) Blockade of BK B2-receptor (Β2R) to prevent BK binding (Figure 3) [69,94,95].
| Drug product | Type | Action | Manufacturer | Approval date | Route | Indication | t1/2b |
|---|---|---|---|---|---|---|---|
| Cinryze® | pd-C1-INH nanofiltered | C1-INH replacement | ViroPharma | 10/2008 | IV | Prophylaxis | 36-48 h |
| Berinert® | pd-C1-INH pasteurized | C1-INH replacement | CSL Behring | 9/2009 | |||
| IV | Acute attacks | 36-48 h | |||||
| Ruconest® | rC1-INH from tg-rabbitsc | C1-INH replacement | Pharming | Under FDA valuationd | IV | Acute attacks | ~ 3 h |
| Kalbitor®(ecallantide) | Recombinant polypeptide | Kallikrein inhibitor | Dyax Corp. | 1/2009 | SC | Acute attacks | ~ 2 h |
| Firazyr®(icatibant) | Synthetic decapeptide | BR B2R antagonist | Shire Orphan Therapies | 8/2011 | SC | Acute attacks | 2-4 h |
Table 2: Therapeutic products recently approved (or under consideration) for treatment of patients with hereditary angioedema in the USAa.
C1-INH replacement therapy
Plasma-derived C1-INH preparations: Since 1973, C1-INH concentrates have been widely used in Europe for treatment of acute attack episodes in patients with HAE [96], but have only recently been FDA-approved for marketing and distribution in the US.
To date, C1-INH replacement therapy for patients with HAE (Table 2) is the only FDA-approved therapeutic application of C1-INH. Two plasma-derived C1-INH (pd-C1-INH) products, Cinryze® and Berinert®, are available in the US for patients with HAE, for prophylaxis and for treatment of acute attacks, respectively (Table 2). The rationale for intravenous (IV) administration of the C1-INH concentrates to restore the levels of functional C1-INH in the circulation is addressing the primary cause of HAE. The human plasma origin of these C1-INH products ensures their tolerability. However, the potential risk of contamination with blood-borne viruses (including unknown and emerging pathogens) still exists in case of protein therapeutics derived from human plasma, despite effective viral clearance steps in the manufacturing procedures [97]. While, in general, the pd-C1-INH preparations are well tolerated, at higher than recommended doses, risk of thromboembolic events may exist [98-100]. The low abundance of C1-INH, limited plasma source per se, and high production cost of C1-INH concentrates from pooled human plasma led to the therapeutic development of recombinant C1-INH.
Recombinant Human C1-INH
Recombinant versions of C1-INH have been under research and development for decades. The gene for human C1-INH was successfully expressed in several hosts [36,101-105]. However, the relatively high yields, stability, and functional activity required for therapeutic-scale production have been achieved only when recombinant human C1-INH (rhC1-INH) has been produced in the milk of transgenic rabbits [106-108]. As an alternative and in addition to pd-C1-INH products, rhC1-INH (Ruconest®) was approved by the European Medicine Agency (EMA) for treatment of HAE in 2010 (Table 2). Currently, Ruconest® (International non-proprietary name: conestat alfa) is available in 30 European countries. The product is not yet approved in the US, as for the biological license application to be considered by the FDA, some additional clinical studies were necessary, recently completed [109,110].
Yet, given the theoretical advantage of being free from blood-borne pathogens (at least human ones) rhC1-INH bears a certain risk of inducing immune response to rabbit milk protein impurities. Whereas the potency of rhC1-INH is comparable to that of pd-C1-INH, due to differences in glycosylation patterns [111], its half-life in circulation is significantly lower than that of pd-product, thus making it more appropriate for treatment of acute attacks (Table 2). Production of therapeutic protein in transgenic rabbits opens up the possibility of a technically scalable and theoretically “unlimited” source of rhC1-INH.
Other drugs for treatment of HAE
Whereas the rationale for C1-INH-based treatment is to restore the plasma level of functional C1-INH, two recently approved compounds (Table 2; Figure 3) reflect treatment approaches other than replacement therapy, namely specific inhibition of plasma kallikrein (ecallantide) and its action by selective blockade of the BK B2-receptors (icatibant). Ecallantide (Kalbitor®) is a small recombinant protein (60 amino acid residues) produced in the yeasts Pichia pastoris; the drug specifically binds and inhibits plasma kallikrein [4,112]. Icatibant (Firazyr®) is a synthetic decapeptide that exhibits a sole specificity to BK B2-receptor (BK B2R); as a selective, competitive antagonist to BK B2R, it blocks the receptor for BK [69,113]. Notably, these two recently approved drugs provide a subcutaneous administration option for the first time (a subcutaneous infusion of C1-INH concentrates for long-term prophylaxis is under active consideration [88]).
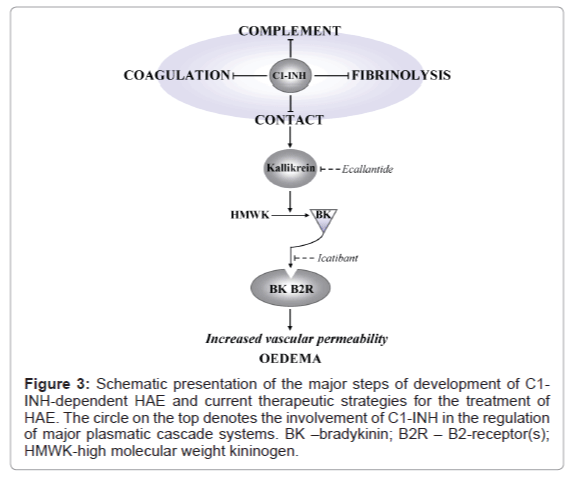
Figure 3: Schematic presentation of the major steps of development of C1- INH-dependent HAE and current therapeutic strategies for the treatment of HAE. The circle on the top denotes the involvement of C1-INH in the regulation of major plasmatic cascade systems. BK –bradykinin; B2R – B2-receptor(s); HMWK-high molecular weight kininogen.
C1-INH potential therapeutic applications other than HAE
As a potent anti-inflammatory agent, C1-INH has also been considered for treatment of several other serious pathological conditions, including sepsis, acute myocardial infarction, vascular leakage syndromes, ischemia-reperfusion injury, brain ischemic injury and some other conditions [7,50,51,114]. Although these applications are promising and seem to be safe, more clinical data are required to support their efficacy.
Current state and future perspectives of C1-INH research and development
Over the last fifty years, tremendous progress in the characterization of C1-INH structure and biological activities has been made. From the standpoint of the biochemistry of C1-INH, many aspects of its structure, potentiation, and interactions with various endogenous ligands still remain to be studied in more detail. In particular, it includes resolution of the crystal structure of the whole protein and elucidation of the role of the N-terminal domain and its excessive glycosylation. The investigation of C1-INH has been tightly linked to and accelerated by the importance of HAE. Not only do we now have a better understanding of C1-INH biochemistry and its therapeutic potential, but recent developments have also brought several licensed C1-INH therapeutic products for replacement therapy in patients with HAE to the US and to the EU. With respect to the recently approved C1-INH products, analyses of long-term safety and efficacy profiles over the coming years in clinical use will be essential. These C1-INH products, together with recently approved small protein drugs (ecallantide and icatibant), drastically changed the available treatment options. Given a high variability in acute attacks sites, frequency, and severity, these therapeutic options offer an opportunity for individualized treatment, selective or complex, specifically tailored for each HAE patient. As there are no head-to-head clinical trials yet, and no data that would allow any comparison between the therapies, future clinical experience is extremely important. From the standpoint of possible enhancement of already existing C1-INH therapies, the potentiation of C1-INH via fine tuning with GAGs is very attractive direction. From the standpoint of further research and development toward using C1-INH for treating conditions other than HAE (such as sepsis or ischemia-reperfusion injury), the next decade certainly will bring novel C1-INH therapeutic applications to light.