Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2015) Volume 3, Issue 2
The Hannover bone marrow (BM) classification distinguished three phenotypes of BCR/ABL-positive CML: CML of common type (CML.CT), CML with megakaryocyte increase (CML.MI) and CML with megakaryocyte predominance (CML.MP). BCR/ABL-positive essential thrombocythemia (Ph-positive ET) is featured by CML.MP bone marrow picture of small monolobulated megakaryocytes and is part of the CML spectrum as a malignant disease (neoplasia) with an obligate transition into acute leukemia of near to 100% after 10 years follow-up. The Hannover BM classification distinguished three primary prefibrotic BCR/ABL-negative (Ph-negative) myeloproiferative disorders (MPD)s: essential thrombocythemia (ET), polycythemia vera (PV) and chronic or primary megakaryocytic granulocytic myeloproliferation (CMGM/PMGM). The incidence of blasts crisis is low in the Phnegative MPDs ET, PV and CMGM. The risk of myelofibrosis is high in CMGM/PMGM, moderate in PV but low in Ph-negative ET. In BCR/ABL-positive thrombocythemia the platelets are small and indolent (non-reactive) and megakaryocytes are smaller than normal with hypolobulated nuclei caused by BCR/ABL induced maturation defect. BCR/ABL-positive thrombocythemia does not present erythromelalgic thrombotic or bleedings manifestations at increased platelet count in excess of 400 to 1500 × 109/L. The platelets and megakaryocytes in BCR/ABL-negative ET and PV are large due to growth advantage caused by constitutively activated by the JAK2V617F or MPL515 mutation. JAK2 and MPL mutated thrombocythemias are associated with a high risk on aspirin responsive plateletmediated inflammation and thrombosis in the end-arterial circulation (platelet thrombophilia).
Keywords: Philadelphia chromosome; BCR/ABL fusion gene; Chronic myeloid leukemia; Essential thrombocythemia; Polycythemia vera; Myelofibrosis; Neoplasia; Myeloproliferative disorders; Myeloproliferative neoplasms
The Philadelphia chromosome is a disease specific marker for CML patients [1]. Rowley discovered that the a large part of chromosome 22q is translocated to 9q resulting in the minute Ph1-chromosome and that a small part of chromosome 9q is translocated to chromosome 22q resulting in the translocation t(9;22) (q34;q11) [2,3]. Heisterkamp. Groffen Grosveld found that c-ABL was translocated to the Ph1-chromosome and subsequently discovered the breakpoint cluster region (BCR) on chromosome 22 [4-7]. The BCR/ABL fusion gene produces a BCR/ABL protein, which has a high tyrosine kinase activity and CML-transformation capacity [8-10]. Ninety percent of all CML patients are Ph1+/BCR/ABL+, 5% are Ph-/BCR/ABL+, and 5% are Ph-/BCR/ABL- , the latter group usually diagnosed as atypical CML, juvenile CML, chronic neutrophilic leukemia or chronic myelomonocytic leukemia [11]. The tyrokinase activity of the BCR/ABL protein is crucial for the leukemic transformation of hematopoietic cells in vitro and in vivo [8-12]. In this report we summarize the clinical presentation and natural history of BCR/ABL-positive CML and natural history in the period before the introduction of imatinib. The development of imatinib as a therapeutic agent for CML became reality in the 21th century changed the prognosis of patients with CML from poor to good [12].
BCR/ABL positive CML is a malignant disease with an obligate transition into acute leukemia, whereas the BRC/ABL negative chronic myeloproliferative disorder (MPD) are featured by a benign neoproliferation of one, two or the three hematopoietic cell lines with a normal or near normal survival in the initial 10 to 15 years of follow-up [13]. Michiels et al and the German pathologists Georgii and Thiele recognized that small mono- or bi-nucleated megakaryocytes are diagnostic for the Ph-positive diseases ET and CML. In contrast, large megakaryocytes with hyper-lobulated nuclei are pathognomonic for Ph-negative essential thrombocythemia (Figures 1 and 2) [13-24]. The Hannover Bone Marrow Classification of CML and MPD regarded myelofibrosis (MF) as a reactive secondary feature of myeloproliferative disease. The terms agnogenic myeloid metaplasia (AMM), myeloid metaplasia with myelofibrosis (MMM), and chronic idiopathic myelofibrosis (CIMF) and primary myelofibrosis (PMF) lack accuracy because it is applied to both the early prefibrotic hypercellular stage and the advanced fibrotic stages [15,16]. Secondary MF in ET, PV and CMGM is classified as reticulin fibrosis (RF) grade 0 and 1 (=MF 0), RF grade 2 (=MF 1), RF 3 with minor collagen fibrosis (=MF 2) and advanced reticulin-collagen fibrosis (RCF) with or without osteosclerosis (=MF 3) [17,18].
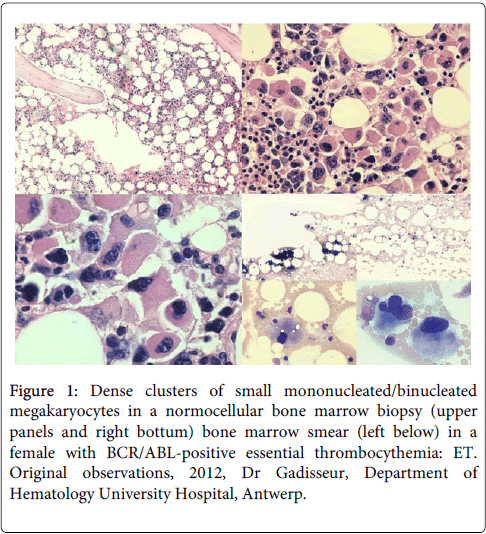
Figure 1: Dense clusters of small mononucleated/binucleated megakaryocytes in a normocellular bone marrow biopsy (upper panels and right bottum) bone marrow smear (left below) in a female with BCR/ABL-positive essential thrombocythemia: ET. Original observations, 2012, Dr Gadisseur, Department of Hematology University Hospital, Antwerp.
In the Hannover Bone Marrow Classification the megakaryocytes in BRC/ABL-positive thrombocythemia and CML are small containing uni- or bi-lobulated nuclei (Figures 1 and 2) but large and mature with hyperlobulated nuclei in ET [16-26]. The megakaryocytes in trilinear PV are of medium size to large (pleomorphic) with hyperploid nuclei. The characteristic increase of clustered enlarged pleomorphic megakaryocytes in a hypercellular bone marrow due to increased erythropoiesis and granulopoiesis (trilinear PV), and no stainable iron are the diagnostic hallmark of PV to distinguish it from congenital and secondary erythrocytoses and reactive thrombocytosis.
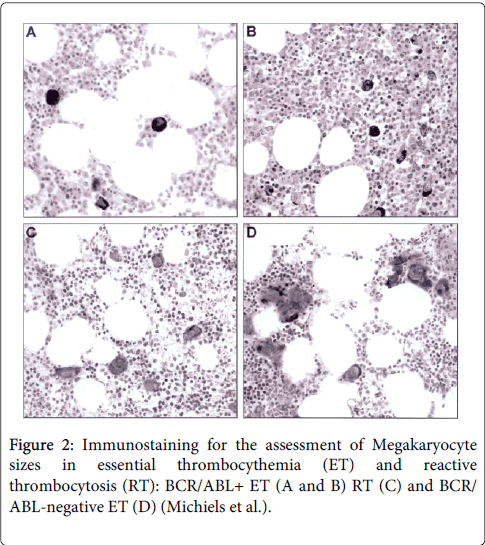
Figure 2: Immunostaining for the assessment of Megakaryocyte sizes in essential thrombocythemia (ET) and reactive thrombocytosis (RT): BCR/ABL+ ET (A and B) RT (C) and BCR/ ABL-negative ET (D) (Michiels et al.).
Prefibrotic CMGM is dominated by primary megakaryocytic-granulocytic myeloproliferation (PMGM) and increase of clustered atypical dysmorphic megakaryocytes due to increases of cellular and nuclear sizes. In CMGM/PMGM, the nuclei of dysmorphic megakaryocytes are bulky with clumsy lobuli and irregular roundish shaped form (so-called cloud-like nuclei), which are never seen in ET, PV and CML [16-26]. CMGM/PMGM is not preceded or followed by normocellular ET, PV or MDS and can be regarded as the third distinct prefibrotic primary MPD (Table 1) [19-29].
| WHO | Textbooks | Hannover Classification | Percentage (%) |
| CML and Subtypes | CML or CGL and Subtypes | Primary diseases CML CML.CT CML.M |
13.9 |
| P.VERA | P.VERA | P.VERA | 17 |
| Thrombocythemia | Thrombocythemia | Thrombocythemia CMGM Pre-early fibrotic |
7.1 16 |
| Agno genic myeloid metaplasia (AMM) | Primary idiopathic myelo fibrosis or agno genic myeloid metaplasia | Advanced Disease Increase of blasts Increase of fibres |
33.3 |
| Unclassifiable | Unclassifiable | Unclassifiable | 12.6 |
| Percentage from 3933-Primary or Idiopathic or Essential Thrombocythemia | |||
Table 1: The hannover bone marrow classification for CMPD.
Georgii et al. distinguished three bone marrow histology types of BCR/ABL-positive chronic myeloid leukemia (CML): CML of common type with a predominance of granulopoiesis (CML.CT), CML with megakaryocyte increase (CML.MI), and CML with megakaryocytes predominance (CML.MP (Figure 3) [16]. CML-MP is rare and usually seen in Ph-positeve ET without features of CML in blood and bone marrow. The incidence frequency ratio of CML.CT versus CML.MI is 3:1. There are differences between CML.CT and CML.MI in their hematologic features as well as in the clinical course. Significant differences were found in platelet counts, which were more than 2-fold higher in CML.MI as compared to CML.CT (574 versus 257 × 109/L). Both reticulin fibrosis (RF) and advanced reticulin-collagen fibrosis (RCF) occurs more frequently in CML.MI than in CML.CT (49% vs. 14%) in 549 diagnostic bone marrow biopsies collected between 1977 and 1987 (Table 2) [18]. Life expectancy did not significantly differ among the subtypes of CML.CT and CML.M. Patients with CML.M with megakaryocyte increase (MI) grade 2 and 3 have a higher risk to develop myelofibrosis as compared to CML.CT with CML.MI grade 0 or 1 (Figure 4) [18].
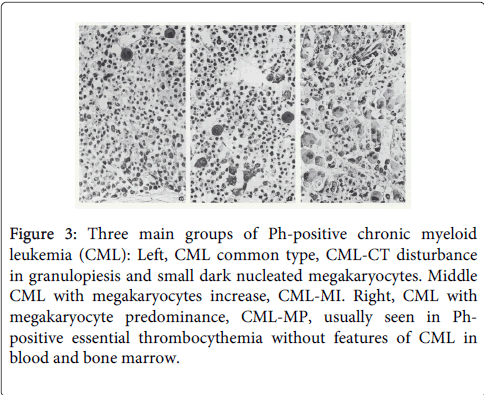
Figure 3: Three main groups of Ph-positive chronic myeloid leukemia (CML): Left, CML common type, CML-CT disturbance in granulopiesis and small dark nucleated megakaryocytes. Middle CML with megakaryocytes increase, CML-MI. Right, CML with megakaryocyte predominance, CML-MP, usually seen in Phpositive essential thrombocythemia without features of CML in blood and bone marrow.
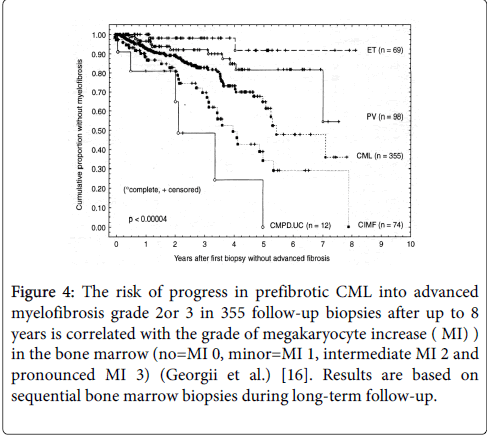
Figure 4: The risk of progress in prefibrotic CML into advanced myelofibrosis grade 2or 3 in 355 follow-up biopsies after up to 8 years is correlated with the grade of megakaryocyte increase ( MI) ) in the bone marrow (no=MI 0, minor=MI 1, intermediate MI 2 and pronounced MI 3) (Georgii et al.) [16]. Results are based on sequential bone marrow biopsies during long-term follow-up.
| Primary Disease (1998) | CML.CT | CML.M | Hannover |
| Number | 415 | 134 | Classification MF |
| 86% | 49% | MF 0=RF 0/1 | |
| 7% | 27% | MF 1=RF 2 | |
| 2% | 20% | MF 2 3=RCF ¾ | |
| RCF=recticulin andcollagen fibrosis | |||
Table 2: Grading of (reticulin fibrosis (RF) and reticulin-collagen fibrosis (RCF) and Hannover classification of myelofibrosis (MF) in 549 diagnostic bone marrows from patients with Ph1+ CML.CT and CML.M collected between 1977-1987 (Georgii et al. [16]).
In the large Hannover study of 1223 CML, 939 ET, 889 PV and 1026 CIMF/CMGM patients myelofibrosis (MF) grade 1 to 3 in diagnostic bone marrow biopsies was 20% in CML, 3% in ET, 12% in PV and 40% in CMGM/CIMF (Table 3) [17]. The development of MF grade 0/1 into grade 2 or 3 in sequential bone marrow biopsies at 7 years after the first diagnostic bone marrow was 10% for ET, 20% for PV and 70% for CMGM/CIMF (Figure 5) [17]. Georgii compared the natural history of BCR/ABL-positive CML with BRCR/ABL-negative-prefibrotic CMGM/PMGM, ( labeled as CIMF in the 2001 WHO or primary myelofibrosis, PMF in the 2008 WHO classification) as detected by blast crisis outcome studies in follow-up bone marrow biopsies [17,18].
| Increase of: Cellularity (increase) |
CML | P.vera | ET | CMGM or CIMF |
| Evident | moderate | Normal | moderate | |
| Granulopoiesis (increase) Blast excess (EB 2-3) Basophils Eiosinophils |
Evident 8.3% Increased increased |
Slight 0.4% Normal varying |
Normal 0.2% Normal increased |
Moderate 1.7% Normal varying |
| Erythropoesis | reduced | increased | normal | Reduced or increased |
| Iron storage | reduced | negative | normal | reduced |
| Megakaryopoiesis Number of MK Cellular size Nuclear size Nuclear lobulation Pleomorphy Clustering |
Varying Reduced Reduced Reduced Low to evident varying |
increased increased increased varying moderate varying |
Increased increased increased increased minimal moderate |
Increased increased increased reduced prominient Eminient |
| Myelofibrosis (MF (1-3)) | 20% | 11.2% | 2.9% | 40% |
| Pseudo Gaucher cells | 70% | Rare | Unusual | unusual |
| Lymphoid nodules | Unusual | Evident | Rare | Rare |
Table 3: Bone marrow (BM) histological features in early stages of chronic myeloproliferative disorders (MPD) in a total of 4167 diagnostic BM biopsies: chronic myeloid leukemia, CML N=1223, polycythemia vera, P.vera N=889, essential thrombocythemia, ET N= 939 and chronic megakaryocytic granulocytic myeloproliferation, CMGM, or chronic idiopathic myelofibrosis, CIMF N=1026 according to Georgii et al. [17].
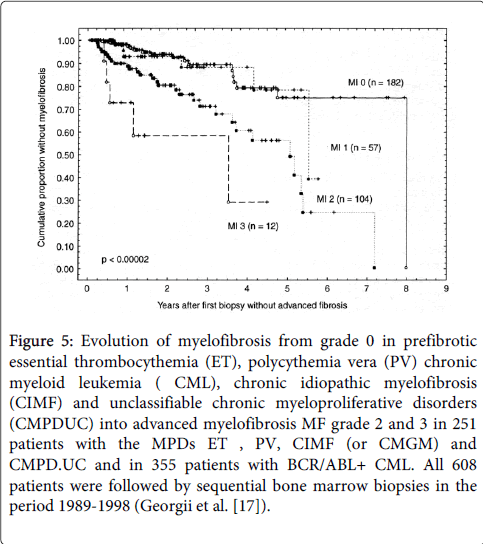
Figure 5: Evolution of myelofibrosis from grade 0 in prefibrotic essential thrombocythemia (ET), polycythemia vera (PV) chronic myeloid leukemia ( CML), chronic idiopathic myelofibrosis (CIMF) and unclassifiable chronic myeloproliferative disorders (CMPDUC) into advanced myelofibrosis MF grade 2 and 3 in 251 patients with the MPDs ET , PV, CIMF (or CMGM) and CMPD.UC and in 355 patients with BCR/ABL+ CML. All 608 patients were followed by sequential bone marrow biopsies in the period 1989-1998 (Georgii et al. [17]).
Table 3: Bone marrow (BM) histological features in early stages of chronic myeloproliferative disorders (MPD) in a total of 4167 diagnostic BM biopsies: chronic myeloid leukemia, CML N=1223, polycythemia vera, P.vera N=889, essential thrombocythemia, ET N= 939 and chronic megakaryocytic granulocytic myeloproliferation, CMGM, or chronic idiopathic myelofibrosis, CIMF N=1026 according to Georgii et al. [17].
In a large 1989-1998 cohort of 1223 CML patients the majority had prefibrotic disease (MF 0 in 78%) and 90% did not have significant excess of blasts in bone marrow biopsies at time of first diagnosis (Table 4) [17]. The incidence of blast crisis (neoplasia) after 8 years follow-up was around 10 to 15% in the cohort of and MPD (ET,PV and CIMF) patients (N=223) but much higher near to 90% in CML (N=315) (Figure 6) [17], clearly consistent with the clinical observations that that CML indeed is featured by an obligate transformation into acute leukemia within 10 years of follow-up as documented in the German prospective CML management studies (Figure 7) [27].
| Number of cases | 1323 | 1323 | |
| Grading MF 199821 | Frequency | Grading EB | Frequency |
| MF-0; No RF; | 78% | EB 0/1 blasts <20% | 90% |
| MF 1 RF 2 | 11% | EB 2Blasts >20% | 5% |
| MF 2 RCF 3/4 | 4% | EB 3Blasts >30% | 3.3% |
| MF3 RCS&O | 4% | - | - |
Table 4: Detection and grading of myelofibrosis (MF) and excess of blasts (EB) in 1323 diagnostic bone marrows from 1323 patients with Ph1+ CML (CML.CT and CML.M) collected between January 1989 and December 1998 (Georgii et al. [17]).
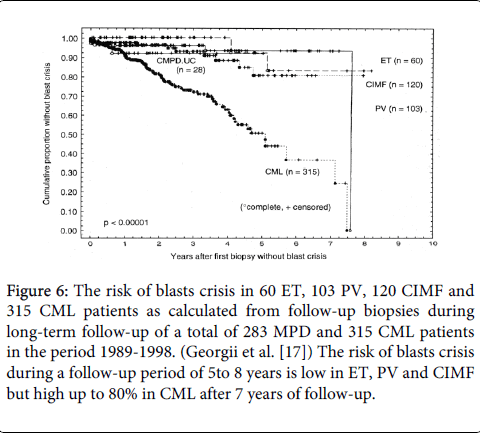
Figure 6: Evolution of myelofibrosis from grade 0 in prefibrotic essential thrombocythemia (ET), polycythemia vera (PV) chronic myeloid leukemia ( CML), chronic idiopathic myelofibrosis (CIMF) and unclassifiable chronic myeloproliferative disorders (CMPDUC) into advanced myelofibrosis MF grade 2 and 3 in 251 patients with the MPDs ET , PV, CIMF (or CMGM) and CMPD.UC and in 355 patients with BCR/ABL+ CML. All 608 patients were followed by sequential bone marrow biopsies in the period 1989-1998 (Georgii et al. [17]).
The poor survival curves in CML patients show some advantage in IFN and HU treated as compared to Busulphan treated CML patients (Figure 7). Variables used in the multivariate step-wise analysis in the Cox model included fiber density RF0/1 versus RF 2/3 in bone marrow biopsy and spleen size ( but not platelet and leukocyte count hemoglobin or blasts percentage in blood or bone marrow) were used to create 3 risk groups CML patients: risk low n=163, intermediate n=167, high n=165. Such retrospective risk stratification has indeed some impact on prognosis, but none of the CML patients survived after 10 years follow-up (Figure 7) [27].
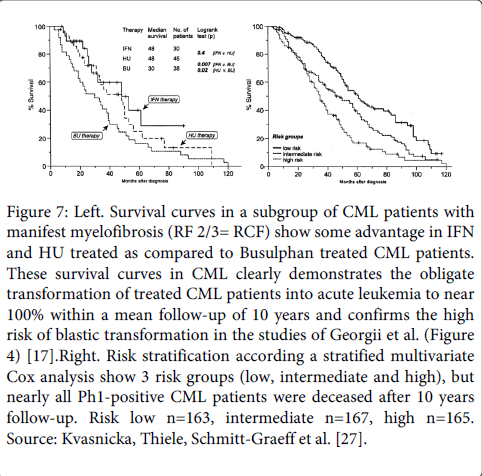
Figure 7: Left. Survival curves in a subgroup of CML patients with manifest myelofibrosis (RF 2/3= RCF) show some advantage in IFN and HU treated as compared to Busulphan treated CML patients. These survival curves in CML clearly demonstrates the obligate transformation of treated CML patients into acute leukemia to near 100% within a mean follow-up of 10 years and confirms the high risk of blastic transformation in the studies of Georgii et al. (Figure 4) [17].Right. Risk stratification according a stratified multivariate Cox analysis show 3 risk groups (low, intermediate and high), but nearly all Ph1-positive CML patients were deceased after 10 years follow-up. Risk low n=163, intermediate n=167, high n=165. Source: Kvasnicka, Thiele, Schmitt-Graeff et al. [27].
A 47 year old female patient with antecedents of a carcinoma of the ovarium 5 years earlier was transferred to the hematology department of the Antwerp University hospital because of a massive thrombocytosis (>5000 × 109/l) in the presence of a normal hemoglobin (16 g/dl) and borderline high leukocyte count (10.6 × 106/l) with a normal formula. The JAK-2 mutation proved negative while BCR-ABL was positive. Bone marrow examination showed a myeloproliferative disease with dense clusters of small megakaryocytes and mono-lobulated nuclei consistent with the diagnosis of BCR/ABL-positive ET (Figure 1). Repeated thrombocyte apheresis and hydroxyurea was initiated (Figure 8). After discontinuation of thrombocyte apheresis the thrombocyte count increased again under hydroxyurea treatment while the leucopenia deepened, and the patient developed a toxic rash. She was then started on Anagrelide (Xagrid®) which resulted in slowly decreasing thrombocyte counts and recuperation of the leucocytes. and a diagnosis of CML was made. Because of development of thrombocytopenia Anagrelide was stopped after 14 days. Imatinib® was started 5 weeks later when thrombocyte counts returned to 4244 × 109/l. Under this treatment the thrombocyte count only decreased slowly over a period of 7 months (Figure 8). Patient compliance proved to be an important problem. After these 7 months there was an evolution from chronic phase CML with high thrombocytosis to an acceleration phase/blast crisis with increased leucocyte counts (141 × 106/l), appearance of peripheral blasts, and thrombocytopenia (117 × 109/l). Imatinib® was switched to second generation TKI (Dasatinib®).
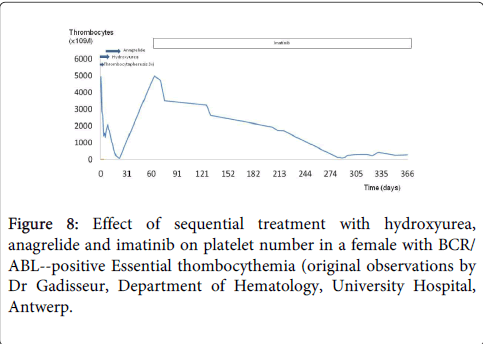
Figure 8: Effect of sequential treatment with hydroxyurea, anagrelide and imatinib on platelet number in a female with BCR/ ABL--positive Essential thombocythemia (original observations by Dr Gadisseur, Department of Hematology, University Hospital, Antwerp.
BCR/ABL-positive ET without features of CML in blood and bone marrow is rare but its presentation and natural history is predicted to be similar to CML.MP. The clinical outcome of BCR/ABL positive ET is poor as compared to BRC/ABL-negative thrombocythemia in MPD [14]. The bone marrow in BCR/ABL positive ET is featured by predominant and pronounced mononucleated megakaryopoiesis with initial no, minor or overt granulocytic hypertrophy consistent with CML.MP (Figure 1). Important differences between BCR/ABL-positive ET and BCR/ABL-positive CML at time of presentation are the predilection of BCR/ABL-positive ET for females and the absence of splenomegaly [14]. Ph-positive ET patients have no features of CML in the peripheral blood at time of presentation. Thrombotic or hemorrhagic events are rare in cases of pronounced thrombocytosis associated with Ph-positive CML during follow-up [28,29]. The prognosis of BCR/ABL-positive ET is rather poor prognosis with the development of CML features, and a high risk of progression to myelofibrosis and blast crisis after a few to several years [13,14]. The bone marrow features of the presented case of Ph-positive ET showed that the megakaryocytes in BCR/ABL-positive thrombocythemia are indeed smaller than normal with hypolobulated round nuclei caused by BCR/ABL gene and protein induced maturation defect of the hematopoietic stem cells (Figure 1). This contrasts with clustered enlarged megakaryocytes in BCR/ABL-negative thrombocythemia in various MPDs (Figure 2) [13,14] due to growth advantage and benign proliferation of constitutively activated JAK2 or MPL somatic mutated megakaryocytes [30-40].
Erythromelalgic thrombotic complications (including migraine-like cerebral or ocular ischemic attacks: MIAs) in ET and PV patients are caused by platelet-mediated inflammation and thrombosis in the end-arterial circulation [29-32]. The platelet-mediated erythromelalgic microvascular thrombotic complications are reversible by aspirin and platelet reduction to normal (<350 × 109/L), but not by coumadin [31,32].
Erythromelalgic thrombotic complications is rare in BRC/ABL positive CML [28,29]. Despite the high platelet count, the presented case of BCR/ABL-positive thrombocythemia did not present neither erythromelalgic microvascular ischemic events nor bleeding complications. In BCR/ABL-positive thrombocythemia, the small and indolent are non-reactive whereas large platelets in thrombocythemia of various MPNs are hypersensitive with clinical evidence of platelet-mediated erthromelalgic microvascular manifestations in ET and PV patients [31,32].
Recently it became evident that the large platelet are produced by large pleomorphic megakaryocytes in patients with ET and PV carrying the JAK2V617F36 or MPL515 mutation [34] JAK2V617F or MPL515 mutated platelets are constitutively activated and hyper-reactive that can readily explain the high risk of platelet-mediated inflammation and thrombosis in the end-arterial circulation in ET and PV patients [34].
Evidence for hypersensitive activated platelets in thrombocythemia stem from our platelet kinetic studies using autologous platelets labeled with 51Cr-sodiumchromate in symptomatic ET and PV patients as compared to asymptomatic patients with reactive thrombocytosis or thrombocytosis associated with Ph-positive CML (Table 5) [29-31].
| Platelet kinetics: 109/L | Platelets | half life time | mean life | maximal life | platelet | |
| Patient | N | 109/L (R) | Time days | Span days | Spandays | turnover |
| Control | 4 | 138-193 | 3.90.3 | 6.91.6 | 8.90.9 | 4620 |
| Group A | ||||||
| Ph1+CML/ RT | 6 | 722-2244 | 4.10.4 | 7.81.2 | 8.90.9 | 203107 |
| Group B | ||||||
| ET, PV | 6 | E- 506-1722 | 3.30.3 | 5.40.7 | 8.00.9 | 335119 |
| Group C | ||||||
| ET, PV | 8 | E+ 489-1130 | 6.6 0.8 | 572374 | ||
Table 5: Platelet kinetic studies in Group A: 4 healthy controls, 6 asymptomatic thrombocytosis including 3 Ph1+ thrombocytosis, 3 reactive thrombocytosis, RT), Group B, 8 with asymptomatic ET or PV, and Group C: 6 with ET or PV complicated by erythromelalgia (compare results with Figure 9). N=normal, R=range, RT=reactive thrombocytosis, CML =chronic myeloid leukemia associated with significant thrombocytosis, ET=essential thrombocythemia, PV= polycythemia vera. E-asymptomatic ET and PV. E+ symptomaticET and PV colicatedby erythromelalgia. Citation: Michiels JJ, Ten Kate FWJ, Raeve HD, Gadisseur A (2015) Bone Marrow Features and Natural History of BCR/ABL-Positive Thrombocythemia and Chronic Myeloid Leukemia Compared to BCR/ABL-Negative Thrombocythemia in Essential Thrombocythemia and Polycythemia Vera. J Hematol Thrombo Dis 3: 192. doi:10.4172/2329-8790.1000192 Page 6 of 9 J Hematol Thrombo Dis ISSN:2329-8790 JHTD, an open access journal Volume 3
Platelet kinetic studies were performed in 3 groups of patients: Group A: 4 healthy controls, 6 asymptomatic thrombocytosis ( 3 Ph1+ thrombocytosis, 3 reactive thrombocytosis, RT), Group B, 8 with asymptomatic ET or PV, and Group C: 6 with ET or PV complicated by erythromelalgia.
The results are summarized in Table 5 and Figure 9. The linear platelet and disappearance survival curves (A, Figure 9, left) in 6 CML/RT patients was associated with normal platelet survival times indicating that platelets in Ph1+ thrombocythemia and RT and leave the circulation by senescence and no platelet consumption.
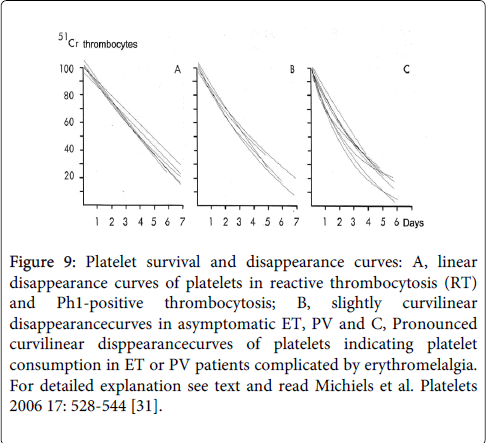
Figure 9: Platelet survival and disappearance curves: A, linear disappearance curves of platelets in reactive thrombocytosis (RT) and Ph1-positive thrombocytosis; B, slightly curvilinear disappearancecurves in asymptomatic ET, PV and C, Pronounced curvilinear disppearancecurves of platelets indicating platelet consumption in ET or PV patients complicated by erythromelalgia. For detailed explanation see text and read Michiels et al. Platelets 2006 17: 528-544 [31].
The concave platelet survival and disappearance curves (C, Figure 9, right) in 8 thrombocythemia patients (ET or PV) with erythromelalgia (E+) are associated with significantly and pronounced shortened platelet survival time (P=<0.01).
The concave platelet disappearance curves (C, Figure 9, right) demonstrates that ET or PV complicated with erythromelalgia and thrombocythemia indeed is featured by platelet consumption caused by arteriolar platelet-mediated thrombosis as could be seen in skin biopsies from erythromelalgic areas of the same patient [31,32].
The shortened platelet survival times in 4 symptomatic E+ patients could be corrected to normal by aspirin 500 mg/day, which was associated with the disappearance of erythromelalgia clinically as well as in skin biopsy [31,32].
The clinical PVSG and the Hannover bone marrow classification clearly separate the Ph/BCR/ABL-positive ET, CML from the Ph/BCR/ABL-negative MPDs ET, PV and CMGM. The BCR/ABL fusion gene produces a BCR/ABL protein with high tyrokinase activity as the cause of Philadelphia chromsome and BCR/ABL positive thrombocythemia and CML. The BCR/ABL+ neoplasia is a distinct neoplastic entity with a spectrum of early and overt manifestations of the chronic stable and accelerated phases of CML and an obligate transformation into acute leukemia [27]. The Hannover Bone Marrow Classification of CML & MPD [16], distinguished two subtypes of Ph-positive CML, the common type (CML.CT) in which granulocytic hypertrophy is predominant, and CML in which megakaryocytic hypertrophy in addition to granulocytic hypertrophy, in bone marrow biopsy is identified. BCR/ABL+ET is a third variant with no features of CML in peripheral blood, with predominant hypertrophy of small megakaryocytes , and with no, minor or overt granulocytic hypertrophy consistent with CML. BCR/ABL positive ET usually progress to CML, and shows a high tendency to myelofibrosis and blastic transformation [13,14].
The Hannover bone marrow (BM) classification clearly defined ET, PV, CMGM and advanced fibrotic MPD at the bone marrow level [16]. Normocellular ET is featured by the presence of large mature hyperlobulated megakaryocytes [16-18,26,27]. This definition of ET in Table 6 is used in the European Clinical and pathological (ECP) classification (Table 6) [21,22,26,27] and in the 2001 and 2008 classification (WHO-ET) [34-39]. WHO defined bone marrow histology in ET is defined as normocellular (cellularity <60%). WHO defined bone marrow histology in JAK2 mutated PV is hypercellular due to increased erythropoiesis (60-80%) or due to trilinear hematopoiesis (70-100%). Prefibrotic chronic, primary or essential megakaryocytic granulocytic myeloproliferation (CMGM/PMGM) is the third distinct MPD entity without features of PV in blood and bone marrow (Table 6). Since the discovery of the JAK2V617F mutation as the cause of trilinear MPN (PV, ET and MF) Michiels et al. extended the ECP (Table 6) by including JAK2V617F mutation screening in the 2007 WHO-ECMP [37] and in the 2015 WHO-Clinical Molecular and Pathological (WHO-CMP) classifications [39,40]. Good evidence exist that the characteristic features of prefibrotic JAK2V617F mutated trilinear myeloproliferative neoplasms (MPN) can be defined as a broad biological continuum of the earliest benign stage of normocellular ET, ET with features of PV (prodromal PV), polycythemia vera (PV), and hypercellular ET due to essential megakaryocytic granulocytic myeloproliferation (EMGM) when the integrated 2015 WHO-CMP) criteria are applied [39,40]. The JAK2V617F positive MPNs ET, PV and EMGM (either heterozygous, hetero-homozygous, homozygous or “trizygous 9p”) represent the classical trilinear MPD with bone marrow features showing a broad spectrum of ET and PV with myeloid metaplasia of the spleen and myelofibrosis during very long-term follow-up [39,40]. A small percentage of JAK2 wild type ET carry the MPL515 mutation, which similar to JAK2V617F thrombocythemia is also associated with a high prevalence of erythromelalgic thrombotic complications including MIAs [40]. Recently we discovered that JAK2/MPL wild type ET and MF carrying the CALR mutation is associated with typical PMGM bone marrow characteristics without features of prodromal or overt PV at diagnosis and during life-long follow-up [40].
| ET Major criteria A1 Persistent platelet count in excess of 400×109/L. A2 Increase and clustering of enlarged mature megakaryocytes in bone marrow biopsy ET Confirmative criteria B1 Presence of large platelets in a peripheral blood smear B2 Absence of any underlying disease for reactive thrombocytosis and normal ESR. B3 No or slight splenomegaly on palpation or scan (<15 cm) ET Exclusion criterion Ph+ chromosome or any other cytogenetic abnormality in blood or bone marrow cells PV Major criteria A1 Raised red cell mass. Male >36 ml/kg, female >32 ml/kg consistent with erythrocytes countabove >6×1012/L A2 Absence of primary or secondary erythrocytosis by clinical and laboratory investigation (a typical PV bone marrow excludes erythrocytosis, Kurnick 1972) A3 Slight, moderate or marked increase in bone marrow biopsy material of: clustered, mature, enlarged pleiomorphic megakaryocytes with hyperlobulated nuclei and moderate to marked increase cellularity of megakaryopoiesis/erythropoiesis or typically trilinear mega-erthro-granulopoiesis. No or presence of reticuline fibers and no collagen fibers (no dry tap) PV Minor criteria B1 Thrombocythemia, persistant increase of platelet >400×109/L B2 Leukocytosis, leucocytecount >10×/L and low erythrocyte sedimentation rate (ESR) B3 Raised leukocyte alkaline phosphatase score >100, absence of fever or infection B4 Splenomegaly on palpation or on isotope/ultrasound scanning A1+ A3 plus one of B establishes PV and excludes any variant of erythrocytosis. |
Table 6: European Clinical and Pathological (ECP) criteria for the diagnosis of essential thrombocythemia (ET), polycythemia vera (PV), and hypercellular ET due to chronic, primary or essential megakaryocytic granulocytic myeloproliferation.