Journal of Pollution Effects & Control
Open Access
ISSN: 2375-4397
ISSN: 2375-4397
Research Article - (2016) Volume 4, Issue 4
Aims: Surfaces of cultural monuments are covered with diverse surface-associated microbial assemblages as a result of the substrata such as limestone that the monuments are constructed with as well as the impacts of various environmental pressures on these perpetually exposed structures over a long period. The aim of this study was to examine and compare the epilithic microbial assemblages on two Maya (i.e. Altun Ha and Xunanthunich) sites located in Belize. Methods and results: High-throughput 454 pyrosequencing approach was utilized to elucidate microbial community assemblages on two monuments. Overall, the taxonomic composition of the epilithic assemblages on both sites revealed the numerical dominance by members of the Proteobacteria at 43% and 36.9%, and the Cyanobacteria at 25.8% and 16.6% on Altun Ha and Xunanthunich, respectively. Comparatively, sequence diversity appeared more pronounced among the epilithic assemblages found on Xunanthunich than those on Altun Ha. Conclusions: The occurrence of relatively different epilithic assemblages on the two sites is probably indicative of the variations in the impacts of weathering and other environmental pressures over a long period on both monuments. Significance and impact of study: The results obtained from this study further emphasize the ecological importance of the delineation and elucidation of the bio-geographical distribution and community diversity of epiphytic assemblages on monuments.
Keywords: Epilithic assemblages; 16S rRNA gene; Cultural monuments
Microbial colonization of cultural monuments and archeological sites have attracted significant scientific attention as a result of the attendant biodeterioration and biocorrosion normally associated with the presence of the surface-associated microbial assemblages on such structures [1-3]. Past studies have suggested a strong linkage between microbial colonization and the subsequent weathering as well as biodeterioration that ensues on the monuments to several factors, including various environmental conditions such as changes in humidity, temperature, wind, light and precipitation [3,4]. Coupled with the fact that majority of these monuments are constructed mainly with such substrata as limestone and marble, with relatively high porosity, making them more susceptible to corrosion by ions and acid rains (due mostly to oxides of sulfur and nitrogen) as well as providing niches for diverse microbial groups [4-7].
Previous studies that have examined the taxonomic composition of epilithic microbial assemblages on monuments utilizing various approaches including culture-based, microscopy, PCR-based molecular methods or 16S rRNA clone libraries have reported diverse representation of bacterial taxa and functional groups [2,8-10]. Majority of the epilithic assemblages examined were found to be numerical dominated by members of the Proteobacteria, Cyanobacteria, Actinobacteria, Acidobacteria, and the Bacteroidetes [2]. However, all of these taxa previously implicated are known to be fast growing, easily cultivable and detectable in the environment. Therefore, there is current need to utilize relatively more stringent molecular strategies, such as the high-throughput next-generation sequencing (NGS) technology, that are able to detect slow growing, less dominant, previously unclassified and potentially novel lineages among the surface-associated assemblages to be examined on the cultural heritage sites.
The purpose of this study was to compare the epilithic microbial assemblages on two Maya sites (i.e. Altun Ha and Xunanthunich) in Belize using the high-throughput 454 pyrosequencing approach. These two Maya sites were targeted for sampling due to their location and accessibility within Belize with a tropical climate that alternate intensely between dry and rainy seasons and an average annual temperature of around 89°F (29°C). Specifically, the Altun Ha site is located in the Belize District about 30 miles north of Belize City and about 6 miles west of the shore of the Caribbean Sea, while Xunanthunich is located over 52 miles away from Belize City, but closer to the city of Belmopan. Therefore, the site’s respective locations should further enrich current understanding of the influences of various environmental factors on the microbial growth on such historical sites, in the Western Hemisphere.
Sample collection and extraction of nucleic acids
Epilithic samples were aseptically collected from the surfaces (upper 1 mm) of two Maya Archaeological sites located about 80 miles apart i.e. Altun Ha (17.7640°N 88.3471°W) and Xunanthunich (17.0839°N 89.1339°W) in Belize (Figure 1A and 1B) as previously described [2]. Briefly, sterile wood sticks padded with cotton wool (Fisherbrand, Fisher Scientific, USA) were gently used to transfer epilithic samples in triplicates into sterile Ziploc bags before placing in several ice packs for storage. Four separate biofilm samples were collected at each monument site, each from the North-and East facing walls as well as the South and West-facing walls in order to reduce the influence of structure orientation [11]. The biofilm samples collected from the surfaces of the structures were mostly gray, brown and black in coloration. These were then pooled together to ensure broad representation of each of the epilithic assemblages prior to extraction of community DNA in the laboratory.

Figure 1: Altun Ha (“Rockstone Pond”) located about 30 miles north of Belize City (A) and Xunantunich (meaning “maiden of the rock” or “stone woman”) in Maya is located over 52 miles away from Belize City (B).
Community DNA was extracted from the filters using FastDNA SPIN Extraction kit (MP Biomedicals, Solon, OH, USA) and eluted in 50 ul of sterile deionized water according to the vendor’s instructions. Determination of DNA quantity was then carried out with a NanoDrop Spectrophotometer (NanoDrop 2000, Thermo Scientific, Delaware, USA). The quality of extracted DNA was further assessed by amplifying with the 16S rRNA universal primer sets, 27F (5’ AGA GTT GTA TCM TGG CTC AG 3’) and 1492R (5’GGT TAC CTT GTT ACG ACT T3’) as previously described [12,13].
High-throughput 16S rRNA gene pyrosequencing
The bacterial community structures in the extracted nucleic acids were assessed by 454 pyrosequencing to target the V1-V3 regions of bacterial 16S rRNA genes uni-directionally using the universal primer 518R (5’ GTA TTA CCG CGG CTG CTG G 3’). Barcodes of between 7 to 11 nucleotides in length were designed and incorporated in front of the PCR primers for sorting of multiple samples within a single 454 FLX Titanium run according to standard methods.
Quality trimming and filtering of low quality sequences
The barcode and the Fusion primers are trimmed before any of the bioinformatics commences. Sequences reads without a barcode or a primer region are dropped and not considered for further analysis. Low quality sequences i.e. those less than 300 base pairs as well as those with less than average quality score (value of 25 or less) are filtered out and deleted [14]. Operational taxonomic units (OTUs) were constructed by comparing them to close relatives via global pairwise alignment [15] to determine their close relatives using the BLASTN system. Chimeras were detected in the sequences that were later omitted for further analysis by using the UCHIME program [16].
Community diversity analysis
The sequences were clustered into OTUs after setting 97% distance limit or cutoff similarity value [16,17]. The OTUs were analyzed for species richness, Shannon Index, Simpson’s (Reciprocal) Index of diversity, species evenness, ACE richness estimate and Chao- 1 richness indicator [18-21]. In order to determine whether total diversity was covered by the numbers of sequences screened, Good’s Library Coverage values were calculated using the equation: C=1- (n/N) × 100, where n is the number of unique OTUs and N is the total number of clones examined as previously described [22,23]. Alpha, beta and gamma diversity calculations were carried out according to Whittaker [24] in addition to rarefaction analysis that was performed to also determine the diversity of the clone libraries using the freeware program by CHUNLAB Bioinformatics Made Easy (CLcommunity version 3.30). Taxon exclusive (XOR) analysis was also carried out based on the taxonomic assignment of sequencing read on the two sets of clones obtained from the epilithic microbial assemblages to reveal those sequences present in one library but absent in the other [25].
Nucleotide sequence accession numbers
The nucleotide sequences obtained were already deposited in the NCBI Sequence Read 157 Archive (SRA) under accession identification number SRP059931.
Within the epilithic microbial assemblages examined, bacterial taxa belonging to the Proteobacteria, Cyanobacteria, Acidobacteria, Bacteriodetes, Chloroflexi, and Actinobacteria were detected on both Maya monuments (Figure 2). Specifically, members of the Proteobacteria and Cyanobacteria were the most dominant, occurring at 43% and 25.8% as well as at 36.9%, and 16.6% on Altun Ha and Xunanthunich, respectively. While members of the Firmicutes, Plantomycetes and Armatimonadetes were found at 2.7%, 1.1% and 1.4% on Xunanthunich, their occurrences on Altun Ha were individually however less than 1%. In contrast, numbers of the Gemmatimonadetes occurred at 1.2% within the assemblages on Altun Ha, but were found to be around 0.82% on Xunanthunich.
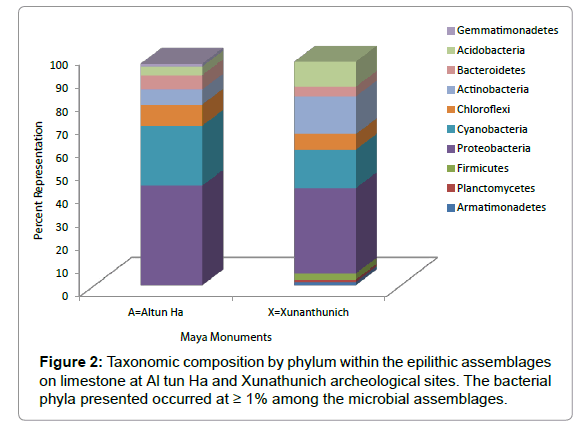
Figure 2: Taxonomic composition by phylum within the epilithic assemblages on limestone at Al tun Ha and Xunathunich archeological sites. The bacterial phyla presented occurred at ≥ 1% among the microbial assemblages.
The rarefaction curve (Figure 3) revealed high levels of bacterial complex within the two epilithic assemblages examined and also showed that neither of the two assemblages had reached the saturation phase, suggesting that the curves might have not totally covered the numbers of different types of bacteria in each of the epilithic communities. However, Good Library Coverage calculation for Altun Ha and Xunanthunich reached 92% and 96%, respectively, a strong indication that majority of total diversity was covered in both epilithic assemblages. Sequence composition analysis based on 97% operational taxonomic units (OTUs) clusters revealed high diversity in the assemblages on the two monuments (Table 1). Specifically, results of bacterial richness and species diversity estimates based on ACE and Chao 1 showed comparatively higher diversity for the assemblages in Altun Ha than Xunanthunich.

Figure 3: Rarefaction analysis of OTUs based on 16S rRNA clone sequences from the epilithic assemblages on limestone at Al tun Ha and Xunathunich archeological sites.
| Monuments | Clone #s | Ace Richness Estimate | Chao1 | Shannon-Wiener index | Non-parametric Shannon | Simpson | Good Library Coverage |
|---|---|---|---|---|---|---|---|
| Altun Ha | 1564 | 3668.49 | 2708.8 | 5.05 | 5.3 | 0.08 | 0.92 |
| Xunanthunich | 1069 | 1557.37 | 1579.77 | 5.07 | 5.19 | 0.04 | 0.96 |
Table 1: Community diversity analysis of the 16S rRNA gene clone sequences from the epilithic assemblages at the 2 Maya archaeological sites in Belize.
When the bacterial assemblages were examined at the class level based on taxon exclusive analysis, the results revealed disparate occurrences of various taxa on the two monuments (Table 2). Eleven bacterial phyla including members of the Chlorophyceae, Mollicutes, Opitutae and Thermoanaerobaculum while present in the assemblages on Altun Ha, were totally absent or undetected on Xunanthunich. Conversely, the epilithic assemblage on Xunanthunich revealed various uncultured bacteria clones, members of the Nitrospira and Elusimicrobia that were not found on Altun Ha.
| Number of 16S rRNA | |||
|---|---|---|---|
| Taxonomic Group | Sequences/Clones | ||
| Class | Phylum | Altun Ha | Xunanthunich |
| Mollicutes | Firmicutes or Tenericutes | 1 | 0 |
| Opitutae | Verrucomicrobia | 16 | 0 |
| Uncultured Bacterium clone GU444092 | - | 1 | 0 |
| Vaucheriales | Chlorophyta | 16 | 0 |
| Chthonomonadetes | Armatimonadetes | 2 | 0 |
| Thermoanaerobaculum | Acidobacteria | 18 | 0 |
| Uncultured Bacterium clone HQ645210 | - | 3 | 0 |
| Fimbriimonadia | Armatimonadetes | 6 | 0 |
| Chlorophyceae | Chlorophyta | 9 | 0 |
| Uncultured Bacterium clone JX016344 | - | 1 | 0 |
| Eustigmatales | Heterokontophyta | 2 | 0 |
| Uncultured Bacterium clone EU289437 | - | 0 | 1 |
| Uncultured Bacterium clone OPB56 | - | 0 | 3 |
| Uncultured Bacterium EF688356 | - | 0 | 1 |
| Uncultured Bacterium clone JF737898 | - | 0 | 3 |
| Nitrospira | Nitrospirae | 0 | 1 |
| Elusimicrobia | - | 0 | 1 |
| Uncultured Bacterium clone DQ404828 | - | 0 | 1 |
Table 2: Result of taxon exclusive analysis at the class/phylum level to detect taxa that are present in one epilithic assemblage but absent in the other based on 16S rRNA gene sequences/clones.
The number of major bacterial phyla (between 8 and 9) that were detected in this study on both Maya monuments examined using the 454 pyrosequencing approach appeared to be relatively higher than previous similar studies that have utilized other approaches such as culture-based, microscopy, PCR-based molecular methods or 16S rRNA clone libraries [2,26,27]. Bacterial phyla including the Proteobacteria, Cyanobacteria, Acidobacteria, Bacteriodetes, and Actinobacteria have been detected previously on Mayan archaeological sites [2] as well as in this study. Specifically, McNamara et al. [2] found members of the Proteobacteria and Actinobacteria to have dominated, representing about 53% and 22% of the 16S rRNA clones respectively, among the epilithic communities from a Maya archaeological site in southern Mexico. Similarly, in this study, the Proteobacteria numerically dominated on both Maya sites examined, with up to 43% and 36.9% abundance, followed by the Cyanobacteria that accounted for 25.8% and 16.6% among the OTUs on Altun Ha and Xunanthunich, respectively. These two bacterial taxa as well as other heterotrophic and photoautotrophic groups have been generally implicated in aesthetic and chemical deterioration of architectural buildings through the formation of biofilms and black crust on the surfaces of such monuments [9,10,28].
However, this present study somewhat differ from some of the previous ones in that more diverse bacterial taxa were implicated on the monuments examined, as compared to the dominance of the Cyanobacteria and easily culturable heterotrophic bacteria that were mostly identified in earlier studies based on the limitation of methodologies utilized [1,26]. In particular, bacterial members belonging to the Plantomycetes, Gemmatimonadetes, Armatimonadetes, Mollicutes, Opitutae and Thermoanaerobaculum were also detected, although at relatively low levels of abundance on the monuments examined in this study. To the best of knowledge, based on available literature, none of these minor constituents of the two assemblages examined in this study have previously been documented on stones associated with archaeological sites. However, previous studies have reported closely related species belonging to the Verrucomicrobia and Chlorophyta phyla to be associated with the biodeterioration of various monumental fountains [10,29].
Comparatively, the taxonomic occurrences and distributions of the major bacterial taxa on the monuments as revealed in this study further corroborates earlier similar studies on Maya archaeological sites that have also documented the dominance of members of the Proteobacteria, Cyanobacteria, Bacteriodetes, and Actinobacteria. These bacterial phyla comprising of diverse nutritional groups including the Photoautotrophs, Chemolithoauthotrophs and Chemoorganotrophs have been observed to be central in the deterioration of monuments in several heritage sites [3]. According to Dakal and Cameotra [3], these bacterial taxa, especially the Cyanobacteria, mostly develop on outdoor monuments due to their simpler nutritional and ecological requirements, including basic inorganic compounds such as atmospheric ammonia, presence of light, carbon dioxide and water that are easily available. Additionally, heterotrophic bacterial populations that have also been associated with monumental stones, includes the Proteobacteria, Actinomycetes and other sulfur-reducing bacterial functional groups, are known to dominate in hypogean environments with characteristic high relative humidity ~90%, low light influxes and constant annual temperature [28,30].
The slight differences observed in microbial taxa occurrences between the two Maya sites, especially in the presence of such taxa as the Mollicutes, Opitutae, Thermoanaerobaculum and Chlorophyceae (presumably as chloroplast genes) on Altun Ha, but totally absent or undetected on those on Xunanthunich could probably be attributed to the orientation of each of the structures, that invariably predisposes each of the Maya sites to variations in the dynamic changes in environmental conditions and microbial growth, over a prolonged period [11]. Overall, the epilithic bacterial populations that were detected within the biofilm assemblages on the surfaces of each of the Maya sites examined may have collectively contributed to the deterioration of these monuments. Several previous studies have attributed the presence and activities of majority of the bacterial taxa detected in this study to the discoloration as well as degradation of stone structures [9,27,31-33]. Therefore, the results from this study further emphasize the ecological importance of delineation and elucidation of the bio-geographical distribution and community diversity of epiphytic assemblages on monuments, especially since the delineation of complex microbial compositions on stone structures might advance effective preservative as well as conservative approaches of such monuments.
This study supported by the Albion College Hewlett-Mellon FDC Funds. Thanks to various staff members of CHUNLAB (Bioinformatics Made Easy, Seoul National University) for assistance during sequencing and analysis of the data. Sincere thanks also to Lori Duff for all the support during the planning stages of the trip to Belize for sampling.