Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2014) Volume 2, Issue 6
Autosomal dominant hereditary Essential Thrombocythemia (ET) due to the gain of function mutation CïG transversion in the splice donor of intron 3 in the TPO gene on chromosome 3q27 in a Dutch and Polish family is associated with marked increased TPO levels and Aspirin-responsive Sticky Platelet Syndrome (SPS). SPS is featured by typical clinical manifestations of aspirin responsive microvascular circulation disturbances including erythromelalgia and atypical transient ischemic attacks. Increase of large platelets in blood smears and large mature megakaryocytes with hyperploid nuclei in a normal cellular bone marrow were diagnostic for autosomal dominant hereditary ET (HET). The spectrum of platelet-mediated thrombophilia in HET is comparable to the aspirin responsive SPS in acquired JAK2V617F positive ET. The affected members of the Dutch and Polish HET families showed no endogenous erythroid colony (EEC) formation. The first generation of the Dutch HET family, two females and one male had stable increased platelet counts, no features of PV, no splenomegaly during life-long follow-up. Three of four elderly family members in the Dutch HET family developed pancytopenia due to myelofibrosis at the age of 71 and 73 years in two, and evolution in acute myeloid leukemia at age 60 in one. These 3 HET patients were on long-term low dose aspirin to prevent SPS manifestations and not treated cytoreductive agents indicating that evolution of ET into myelofibrosis (MF) and leukemia belong to the natural history of TPO-induced HET. In the congenital HET caused by gain of function mutation in the TPO and the JAK2 gene ( JAK2V617I and JAK2R564Q) the responses of mutated CD33 and CD34+ cells to TPO are increased, but the responses to EPO were normal thereby explaining why HET caused by heterozygous germline TPO and JAK2 mutations are associated with the biological characteristics of ET without PV features.
Keywords: Autosomal dominant hereditary essential thrombocythemia; Myeloproliferative neoplasm; WHO classification; JAK2 mutation; TPO gene mutation; Myelofibrosis
In the 1990s, studies on murine leukemia and oncogenes led to the recognition of a new member of the hematopoietin receptor super family [1-5]. The murine myeloproliferative leukemia (MPL) gene is the normal cellular homologue of the oncogene v-MPL and responsible for a panmyeloid transformation capacity [1]. This was followed by the molecular cloning and charaterisation of MPL, the human homologue of the c- and v-MPL [2-4]. The receptor MPL was then rapidly recognized as being the thrombopoietin receptor (TpoR) by the demonstration that antisense oligonucleotides of c-MPL inhibited the colony-forming of megakaryocyte progenitors by Wendling et al. [5]. The MPL ligand became the key to the identification of TPO and was cloned in 1994 by five independent groups [6-10]. The MPL ligand is identical to thrombopoietin and labeled as megakaryocyte growth and development factor (MDGF) [9-12]. Human TPO has all the functions ascribed to MDGF, and all MDGF-like activity can be neutralized by soluble recombinant MPL [13]. TPO stimulates hematopoietic stem cells and megakaryocyte precursors to proliferate, differentiate and mature and mature megakaryocytes form pro-platelets, which then disintegrate into platelets (Figure 1) [13].
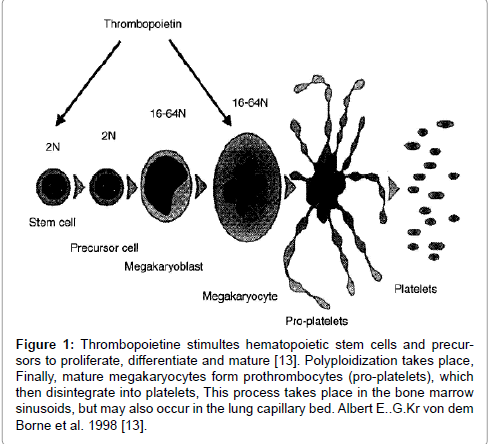
Figure 1: Thrombopoietine stimultes hematopoietic stem cells and precursors to proliferate, differentiate and mature [13]. Polyploidization takes place, Finally, mature megakaryocytes form prothrombocytes (pro-platelets), which then disintegrate into platelets, This process takes place in the bone marrow sinusoids, but may also occur in the lung capillary bed. Albert E..G.Kr von dem Borne et al. 1998 [13].
Hereditary Essential Thrombocythemia (HET): Personal Experiences
The propositus of the Dutch family with hereditary essential thrombocythemia (HET) presented in 1986 with typical erythromelalgia complicated by acrocyanosis of a few toes followed by gangrene and amputation of toe (Table 1) [14,15]. Recurrent erythromelalgia and acrocyanosis in 1986 typically responded to low dose aspirin but not to vitamin K antagonist Aspirin responsive platelet-mediated microvascular disturbances or Sticky platelet syndrome (SPS) [16] was first described by Michiels et al. one year before in 1985 [17] in patients with essential thrombocythemia (ET) and polycythemia vera (PV).The histopathological findings in bone marrow biopsies of the propositus (case II 3, Figure 2) in the Dutch family with HET was compatible with ET very similar to the bone marrow findings in our patients with acquired ET) and PV complicated by erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia [17].
| Clinical data of Dutch HET patients and non-HET subjects | HET (%) | No HET (%) |
| Number of cases | 10 | 5 |
| Age years, range | 11-58 | |
| Vaso-occlusive symptoms: erythromelalgia (E) | 50 | 0 |
| Early E, tip paresthesia | 30 | 0 |
| Red congested erythromelalgia | 10 | 0 |
| E complicated by cold tip feeling-arocyanosis/gangrene | 20/10 | 0/0 |
| Transient ischemic attack: TIA | 30 | 0 |
| Angina pectoris | 20 | 0 |
| Bone marrow histology data of ten HET patiens [14] | (%) | |
| Megakaryopoiesis -increased | 100 | |
| Megakaryocyte clustering of 2-4 cells | 100 | |
| Erythropoiesis increased | 10 | |
| Reticulin-content increased grade 1 | 60 | |
| Storage iron present | 90 | |
| Chromosome abnormalities | 0 |
Table 1: Clinical manifestations and main bone marrow histology data in the Dutch family with Sticky Platelet Syndrome in autosomal hereditary essential thrombocythemia (HET) [14,15].
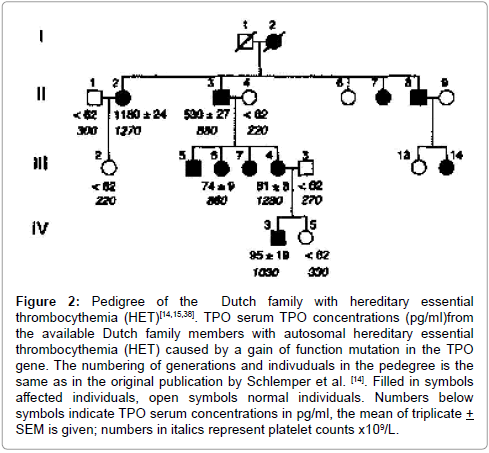
Figure 2: Pedigree of the Dutch family with hereditary essential thrombocythemia (HET)[14,15,38]. TPO serum TPO concentrations (pg/ml)from the available Dutch family members with autosomal hereditary essential thrombocythemia (HET) caused by a gain of function mutation in the TPO gene. The numbering of generations and indivuduals in the pedegree is the same as in the original publication by Schlemper et al. [14]. Filled in symbols affected individuals, open symbols normal individuals. Numbers below symbols indicate TPO serum concentrations in pg/ml, the mean of triplicate + SEM is given; numbers in italics represent platelet counts x109/L.
The propositus case II3 of the Dutch HET family was referred to Dr Michiels at the Erasmus University Medical Center (Academic Hospital Dijkzigt Rotterdam) for expert evaluation. At time of diagnosis of familial ET the histopathology from bone marrow biopsy material from the propositus in 1986 and in 1991 was diagnostic for ET as described by Michiels et al. [17] (Figures 3-5). All features according to the Rotterdam Clinical and Pathological (RCP) diagnostic criteria for ET [17] were present:
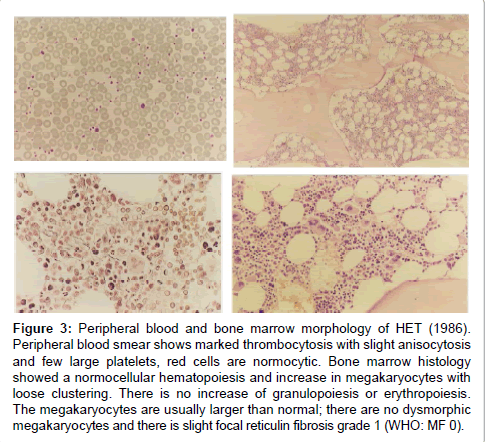
Figure 3: Peripheral blood and bone marrow morphology of HET (1986). Peripheral blood smear shows marked thrombocytosis with slight anisocytosis and few large platelets, red cells are normocytic. Bone marrow histology showed a normocellular hematopoiesis and increase in megakaryocytes with loose clustering. There is no increase of granulopoiesis or erythropoiesis. The megakaryocytes are usually larger than normal; there are no dysmorphic megakaryocytes and there is slight focal reticulin fibrosis grade 1 (WHO: MF 0).
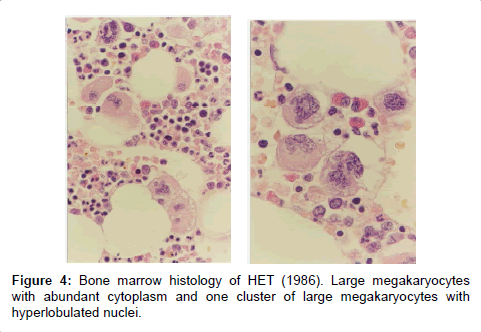
Figure 4: Bone marrow histology of HET (1986). Large megakaryocytes with abundant cytoplasm and one cluster of large megakaryocytes with hyperlobulated nuclei.
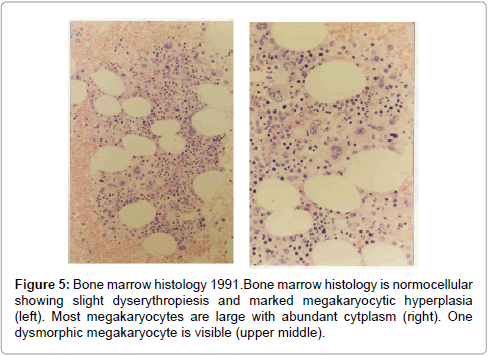
Figure 5: Bone marrow histology 1991.Bone marrow histology is normocellular showing slight dyserythropiesis and marked megakaryocytic hyperplasia (left). Most megakaryocytes are large with abundant cytplasm (right). One dysmorphic megakaryocyte is visible (upper middle).
1. Increase of platelet count in excess of 400 x109/l in the absence of any cause or sign of reactive thrombocytosis (Figure 3).
2. Typically clustering and increase of enlarged megakaryocytes showing mature cytoplasm and hyperploid nuclei in a normocellular bone marrow (Figures 4 and 5).
3. No preceding or allied other subtype of myeloproliferative disorder (MPD) or myelodysplastic syndrome (MDS).
4. Normal cellularity of the bone marrow with only slight increase of fine reticulin fibers.
Based on the publications in the 1990s on the discovery of thrombopoietin (TPO) and its receptor MPL (= TpoR) on megakaryocytes and platelets [1-13]. Wiestner et al. [15] discovered in 1998 that the Dutch HET was caused by a gain of function mutation in the thrombopoietine (TPO) gene with the co-segregation of G to C in the splice donor site of intron 3 in the TPO gene. This gain of function mutation in the TPO gene caused increased TPO levels and microvascular manifestations of affected member of the Dutch HET family (Figure 2 and Table 1, Dutch HET pedigree) [14,15]. While on low dose aspirin since 1986 according to the Rotterdam recommendation in 1985 by Michiels et al. [17], all affected HET patients in the Dutch family were free of microvascular ischemic circulation disturbances and major thrombosis by treatment with low dose aspirin at platelet count between 400x109/L and around 1000x109/L during life long follow up without the need of myelosuppressive treatment. Kondo et al. [18] independently described a second HET family and Liu et al. [19] a third HET family with a similar gain of function mutation in the thrombopoietine (TPO) gene. The laboratory, clinical andbone marrow features of the 11 affected family members of the Polish HET family are summarized in Table of Figure 6. The serum TPO levels in most affected members were increased or even in the upper range of normal. Five of eleven affected family members had thrombocythemia related symptoms including headaches, acrocyanosis, limb parestahesias, venous thrombosis, transient ischemic attacks, miscarriage and thrombo-angiitis obliterans consistent with SPS similar as has been described in acquired thrombocythemia in ET and PV by Michiels et al. [17]. SPS typically responds to low dose aspirin (75 mg/day) and attempts to relieve microvascular ischemic symptoms by cytoreductive therapy with hydroxyurea in the proposita (PL09) were ineffective. The bone marrow histology features in 6 affected members of the Polish HET family were similar to early pefibrotic stage of MPN: increase and loose to dense clusteringas reflected by normal myeloid to erythroid ratio of bone marrow nucleated cells of normal to medium sized mature megakaryocytes with normal to slight increased cellularity according to age, no increase of erythropoiesis, no increase of reticulin fibrosis (RF grade 0 to 1). As compared to controls, the clustered megakaryocytes were more compact, of normal to increased size with slightly hyperlobulated nuclei, but less pronounced as compared to JAK2V617F mutated acquired normocellular ET and prodromal PV (Figure 6).
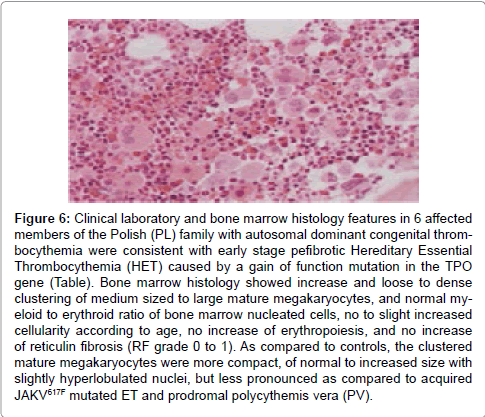
Figure 6: Clinical laboratory and bone marrow histology features in 6 affected members of the Polish (PL) family with autosomal dominant congenital thrombocythemia were consistent with early stage pefibrotic Hereditary Essential Thrombocythemia (HET) caused by a gain of function mutation in the TPO gene (Table). Bone marrow histology showed increase and loose to dense clustering of medium sized to large mature megakaryocytes, and normal myeloid to erythroid ratio of bone marrow nucleated cells, no to slight increased cellularity according to age, no increase of erythropoiesis, and no increase of reticulin fibrosis (RF grade 0 to 1). As compared to controls, the clustered mature megakaryocytes were more compact, of normal to increased size with slightly hyperlobulated nuclei, but less pronounced as compared to acquired JAKV617F mutated ET and prodromal polycythemis vera (PV).
Molecular Etiology and Pathophysiology of TPOInduced Autosomal Dominant HET
Analysis of the TPO gene in his Dutch family with an autosomal inherited ET was initiated by us and performed by Dr Skoda [15]. The CG transversion in the splice donor of intron 3 of the TPO gene cosegregated with the affected autosomal dominant hereditary ET (HET) in the family. This mutation destroys the splice donor site in intron 3 and results in exon 3 skipping. The shortened 5’UTR gene resulted in a gain of function which leads to overproduction of thrombopoietin (TPO) by a mechanism of increased efficiency of the TPO mRNA translation. The in vivo increased TPO levels are responsible for the etiology of hereditary ET by stimulating megakaryocyte production both in vitro and in vivo. Size, number, and mean geometric ploidy of megakaryocytes are increased by TPO as compared with other cytokines with hematopoietic activity. Evidence for a decisive role of deregulated TPO in ET comes from observations in mice overexpressing a TPO transgene where increased TPO production resulted in a fatal myeloproliferative disorder [20]. Lethally irradiated mice grafted with bone marrow cells infected with a retrovirus carrying the murin TPO cDNA induced high TPO levels in mice (TPOhigh mice), who developed thrombocythemia followed by a lethal myeloproliferative disorder of megakaryocytic granulocytic myeloproliferation (MGM) with reduced erythropoiesis in the spleen and bone marrow [21]. In this study, platelets of normal size and morphology in control mice (A) and large platelets in TPOhigh mice were observed initially at time of the ET picture [21]. The continuous forced expression of TPO, (TPOhigh mice) in mice induces megakaryocyte proliferation and differentiation and subsequently develop myelofibrosis [21,22]. TPOhigh mice engineered to overexpress TPO in their liver and those that received transplants of marrow cells infected with a TPO containing retrovirus develop thrombocythemia due to massive hyperplasia of megakaryocytes and granulocytes and hypoplasia of erythropoiesis in the bone marrow followed by myelofibrosis and extramedullary hematopoiesis within 2 to 3 months and die from myeloid metaplasia and myelofibrosis thereafter [21]. TGF-Beta-1 has been implicated in the pathobiology of myelofibrosis by the observation that megakaryocytes from TPOhigh rats and mice express high levels of TGF-Beta- in marrow extracellular fluids and plasma [22]. Another growth factor produced by megakaryocytes, platelet derived growth factor (PDGF), was found to be upregulated in a fashion similar to TGF-Beta1. High levels of TGF-Beta1 mRNA in bone marrow and spleen cells in TPOhighmice were associated with high levels of TGF-beta1 protein in extracellular fluids from these organs [22]. Evolution of ET into transient myelofibrosis has been observed in rats receiving recombinant TPO [21]. Mice respond to TPO treatment by increasing the number of platelets in the circulation and megakaryocytes in the spleen at day 7 to 10 and returned to pretreatment values at day 14 [22]. In wild type mice, TPO treatment increases platelet counts 2.3 fold and the number of CFU-megakaryocytes. TPO treatment had profound effects on the morphology of megakaryocytes in wild type mice. The overall morphology of the megakaryocytes in the spleen became less mature as revealed by reduced localization of P-selectin and von Willebrand factor on the alpha granules. In addition, a significant portion of these megakaryocytes had heavy-electron dense para-apoptotic morphology and contained neutrophils embedded in the cytoplasm, as confirmed by myeloperoxidase immunostaining. In wild mice TPO treatment decreased GATA-1 content in megakaryocytes, and the development of myelofibrosis is associated with high levels of transforming growth factor beta-1 (TGF-Beta-1) expression in bone marrow and spleen [22].
Congenital gain of function mutation in the TPO gene on chromosome 3q27 results in increased levels of plasma TPO levels, which induce a physiological activation of the TPO→MPL signalling pathway. This results in hyperproliferation of large mature megakaryocytes (Figure 2) and increased platelet count complicated by platelet-mediated microvascular complications (Table 1). Platelets do contain many constituents of the TPO signalling machinery. Apart from MPL, platelets contain JAK2-PI-3 kinase, Stat-3 and Stat-5 p38, MAPK and signalling elements downstream of those regulars [23-27]. Platelet stimulation with a supra-physiological plasma concentration of TPO initiates aggregation and secretion, illustrating that TPO act as an independent inducer of platelet responses [28-30]. Pre-incubation of platelet with 20 ng/ml TPO for 5 minutes increases the amount of serotonin secretion by low a low dose of the agonist thrombin (0.1 U/ mL) [23]. Activation of platelets induced by TPO and thrombin leads to the subsequent activation of the enzyme cPLA2, which liberates arachidonic acid from membrane phospholipids, which is the source for platelet cyco-oxygenase (COX-1), thereby causing spontaneous platelet aggregation with formation of platelet thrombi in the endarterial circulation at places of high shear rate consistent with aspirin responsive platelet sticky syndrome (SPS) [17,23,30-33]. In addition to the activation of platelets free in suspension (plasma) by soluble agonist such as thrombin, platelets bind to each other mediated by the von Willebrand factor (VWF) as well as to subendotheliam and specific surface bound VWF protein that become accessible in the damaged vessel wall [23]. Perfusion models mimicking platelet adhesion in flowing blood show that platelets (including VWF-platelet aggregates) bind to these surface-bound adhesive protein in the absence of a soluble platelet activator. Very low concentrations of plasma TPO (0.01 to 1.0 ng/mL) enhance this platelet-VWF adhesion and VWFplatelet aggregation by more than 50%. Adhesion to VWF is preceded by a rolling phase until the interaction with VWF is sufficiently strong. In the presence of 1 ng/mL TPO, firm platelet attachement to subendothelial surface is almost immediate. Subsequently, the formation of circulating VWF-platelet aggregates/thrombi mediated by the high shear stress occurs, thereby causing platelet thrombi attached to the subendothelium appears to be very characteristic for SPS [23]. These findings show that increased plasma TPO concentrations causes hypersensitive platelets in circulating plasma and thereby have a major effect on the initiation of the clinical manifestations of spontaneous platelet-mediated arteriolar inflammation and thrombosis at high shear rate in the end-arterial circulation including aspirin erythromelalgia and its ischemic complications [17] or Sticky Platelet Syndrome16, which is preventable by inhibition of platelet cyclo-oxygenase (COX- 1) by low dose aspirin but not by Coumadin (vitamin K antagonist, Table 1). The clinical picture of aspirin responsive SPS in HET is similar as has been described in 1984/1985 for aspirin responsive erythromelalgia caused by arteriolar inflammation and thrombosis in the endarteriolar circulation in myelopoliferative thrombocythemia of ET and PV patients [17]. Since the discovery of the molecular etiology of myeloproliferative neoplasms (MPN), the association of aspirin responsive SPS (ASPS) and ET could be confirmed to occur in JAK2V617F positive ET, PV, MPL and calreticulin (CALR) mutated ET as well [34,35]. Briefly, aspirin responsive microvascular episodes of fainting and dizziness typical for ASPS were the predominant clinical feature in 23 affected family members of the Dutch and Polish HET families. Disease onset in HEToccur already in childhood in patients with HET and in adults or eldrly patients in acquired ET and PV. The nature and frequencies of aspirin responsive erythromelalgic thrombotic vascular manifestations and hemorrhagic events were similar in congenital HET and acquired ET [17,34].
Natural History of TPO-Induced Autosomal Dominant HET in the Dutch Family
The initial bone marrows histologic findings in the Dutch HET family were blindly and independently evaluated in 2014 by Drs Piche and De Raeve directly from the pictures without any knowledge of the clinical and laboratory data and not aware of the diagnosis of congenital familial thrombocythemia caused by a gain of function mutation in the TPO gene. The first bone marrow biopsy in 1986 of case II3 is consistent with ET according to the 2014 WHO clinical molecular and pathological (2014 WHO-CMP, Figures 3 and 4) [35]. A second bone marrow biopsy in 1991 showed dense clustering of large megakaryocytes megakaryocytes (Figure 5). Bone marrow cellularity is normocellular to slightly increased showing slight dyserythropiesis and marked megakaryocytic hyperplasia. Most megakaryocytes are large with abundant cytplasm with slightly increased reticulin stain grade 1 (Figure 5). The histology picture of the third follow-up bone marrow biopsy in 1996 was dramatically changed (Figures 7 and 8) and showed a hypocllular bone marrow with a few focal dense clustered dysplastic megakaryocytes and reticulin grade 3 to 4 according to PVSG criteria Ellis et al. [36]. The propositus of the Dutch family with the gain of function mutation in the TPO gene showed no further increase of platelet counts, no features of PV, no splenomegaly and ultimately developed end-stage myelofibrosis at the age of 73 years (Figures 7 and 8).
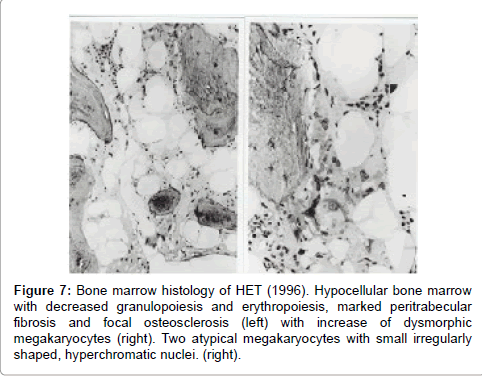
Figure 7: Bone marrow histology of HET (1996). Hypocellular bone marrow with decreased granulopoiesis and erythropoiesis, marked peritrabecular fibrosis and focal osteosclerosis (left) with increase of dysmorphic megakaryocytes (right). Two atypical megakaryocytes with small irregularly shaped, hyperchromatic nuclei. (right).
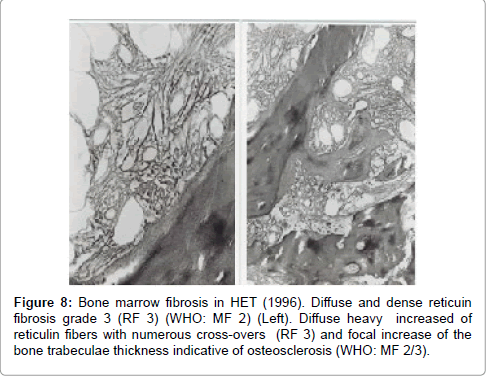
Figure 8: Bone marrow fibrosis in HET (1996). Diffuse and dense reticuin fibrosis grade 3 (RF 3) (WHO: MF 2) (Left). Diffuse heavy increased of reticulin fibers with numerous cross-overs (RF 3) and focal increase of the bone trabeculae thickness indicative of osteosclerosis (WHO: MF 2/3).
Bone marrow histology in 4 affected cases at ages between 15 and 22 years in the third generation of the Polish family showed a slightly hypercellular bone marrow due to increased megakaryopoiesis of loosely clustered small, medium and some large sized megakaryocytes and moderate reactive increase of dispersed mature eosinophils (Figure 6) [19]. The two adult cases in the second generation of the Polish HET family had increase of megakaryopoiesis in a completely normocellular bone marrow at ages of 52 and 66 years indicating the absence of bone marrow hypercellularity similar as in the proband of the Dutch HET case II3 at age 62 years (Figures 3 and 4). All affected members in the Polish HET family had normal bone marrow myeloid/erythroid cells ratios. whereas the myeloid/erythroid ratio was decreased due to an increased erthropoiesis in acquired ET caused by the JAK2V617F mutation (Table in Figure 6) [19].
In 2010 Posthuma et al. updated the natural history of case II2 and II8 of the second generation of the Dutch HET family [37]. Case II 2 of the Dutch HET family died at the age of 71 due to myelofibrosis with severe pancytopenia. Her bone marrow showed myelofibrosis with dysplastic megakaryopoiesis, granulopoiesis and erythropoiesis, and increase of blasts (10%). The peripheral blood showed leukoerythroblastosis, macrothrombocytes, and teardrop cells. She died 3 months after diagnosis of myelofibrosis. Case II8 of the Dutch HET pedigree had a history of diabetes, hypertension and transient ischemic attack in 1989, and was referred in 2008 because of fatigue, anemia and fever (hemoglobin 6.1 mmol/L,leukocytes 4.8x109/L, platelets 90x109/L, 4% blasts and increased LDH, 1509 U/L [37]. Bone marrow cytology showed 45% myeloid blasts (CD34/117/13/33 and HLA-DR positive) and complex cytogenetic abnormalities: 47, add(2) (p2?3, del 5q, i(8((q10) + i (*)(10), -18, -20, i(21)(q10), =2-5 (cp5), and no JAK2V617F mutation. The diagnosis was consistent with AML with maturation not otherwise classifiable according to WHO criteria. The AML was refractory to treatment and the patient died at the age of 71 years [37]. Patients II2, II8 and III3 of the Dutch HET family were treated with low dose aspirin to effectively prevent microvascular ischemic attacks including erythromelalgia and did not receive cytoreductive agents [17,37]. Consequently, the evolution of normocellular ET into myelofibrosis or AML in the second generation at ages around 70 years of the Dutch HET family is part of the natural history of polyclonal TPOinduced HET caused by a gain of function mutation in the TPO gene. Increased plasma TPO levels produced by liver cells caused by a gain of function mutation in the TPO gene on chromosome 3q27 in the Dutch and Polish HET ffamilies do not affect the erythroid and granulocytic hematopoietic stem cells but selectively induced proliferation and differentiation of polyclonal megakaryopoiesis and platelets leading to the disease called hereditary essential thrombocythemia (HET), which after life-long follow-up evolved into pancytopenia associated with myelofibrosis or blastic transformation of hematopietic stem cells at the age around 70 years in the Dutch HET family.
Pathophysiology of Clinical and Bone Marrow Features in Congenital TPO-Induced HET
The frequencies of aspirin sensitive microvascular circulation disturbances (Sticky Platelet Syndrome [16]) were similar in the Dutch and Polish HET families. Platelet counts were above 400x109/L to around 1000x109/L was associated with normal leukocyte and erythrocyte counts, no or slight splenomegaly, and increase of clustered large megakaryocytes in a normocellular bone marrow with normal myeloloid/erythroid ratio and absence of EEC [34-36]. Kralovics et al. compared cMPL expression in the acquired MPDs ET andPV versus HET [38]. He examined 44 patients with MPD (23 PV, 15 ET, and 6 MF) and 18 healthy individuals. Decreased expression of c-MPL protein was found in 30% of patients with PV (7 of 23), 40% of ET (6 of 15) and 67% of IMF (4 of 6). Thus, c-MPL cannot be used as a diagnostic test for PV [38]. To assess whether lower expression of c-MPL is specific for MPD Kralovics et al. studied the Dutch HET family in which thrombocythemia is caused by elevated TPO serum levels due to a splice donor mutation in the TPO gene38. They found lower expression of c-MPL protein in 7 of 8 affected HET individuals (88%), despite normal c-MPL mRNA levels. These findings could be confirmed in the Polish HET family (Table in Figure 6) [19]. Hence, decrease of c-MPL protein also can occur in patients who display sustained monolinear thrombocythemias (HET) caused by a molecular mechanism different from sporadic MPD. Importantly, EECs were negative in all affected Dutch and Polish family members indicating the absence of PV features at the biological bone marrow level. EECs remain the most reliable auxiliary diagnostic assay for PV.
Liu et al. [19] analysed in collaboration with the molecular genetic laboratory of Dr Skoda the large Polish and Dutch families with HET caused by the identical mutation C→G transversion in the splice donor of intron 3 of the THPO gene in 11 affected family members with autosomal dominant HET [19]. The megakaryocytes were normal medium sized enlarged and strikingly compact and clustered in the Polish families with TPO-induced HET caused by the gain of function mutation in the TPO gene (Figure 6). The size of megakaryocytes and the compactness of their nuclei was significantly higher in HET than in normal controls. Similar clinical, laboratory and findings were found in acquired ET at that time [17], and recently also in normocellular JAK2V617F mutated ET [35]. The TPO receptor (TpoR=MPL) protein expression in platelets were down regulated and decreased in the Polish and Dutch HET families (Figure 9). The absence of EEC in the Dutch and Polish HET families indicates the the erythropoietine receptor (EpoR) receptor and its functional status were normal and not influenced or activated by the gain of function mutation in the TPO gene [19,38]. The decreased MPL expression was associated with increased MPL mRNA expression in platelets indicating increased MPL receptor protein turn-over metabolism (Figure 9). From these basic research studies it can be concluded that increased levels of TPO in HET patients are caused by a gain of function mutation in the TPO gene, which selectively activates the MPL (TpoR) pathway and not the EPO/JAK2 pathway thereby causing ET features with increased platelet counts and increase of abnormal polyclonal megakaryocytes arising from normal polyclonal hematopoietic hematopoietic stem cells. The morphology of large polyclonal megakaryocytes with hyperlobulated nuclei in HET caused by the gain of function mutation in the TPO gene isquiete similar to the large megakaryocytes in clonal megakaryopoiesis of patients with acquired heterozygous JAK2V617F mutated ET are quite similar [17,35]. Bone marrow histology in acquired ET complicated by aspirin responsive erythromelalgia typically showed increase of clustered large mature pleomorphic megakaryocytes with normal (<60%, WHO-ET) to increased cellularity (60-80%, prodromal PV) due to pronounced increased erytropoiesis [17,35]. EEC and increased erythropoiesis in the bone marrow typical for PV were not seen in the affected members of the Dutch and Polish HET patients caused by the gain of function mutation in the TPO gene (Tables 1 and 2 in Figure 6). In a separate study we analysed the clinical and hematological features in 41 patients of seven Italian families, including 21 ET patients with a proven MPLS505N mutation and 20 relatives with thrombocythemia reported in the medical records in view of the clinical presentation in 30 ET patients with acquired MPL515 mutation (9 males and 21 females, age 22-84, mean 56 years) [39]. There are at least four distinct variants of acquired ET in terms of molecular etiology: acquired clonal heterozygous JAK2V617F positive ET, JAK2 wild type clonal ET carrying one of the MPL515 or (CALR) mutations [35,39]. All molecular variants of acquired clonal ET usually present with aspirin-responsive platelet sticky syndrome (ASPS) and frequently show evolution into myelofibrosis [35,40].
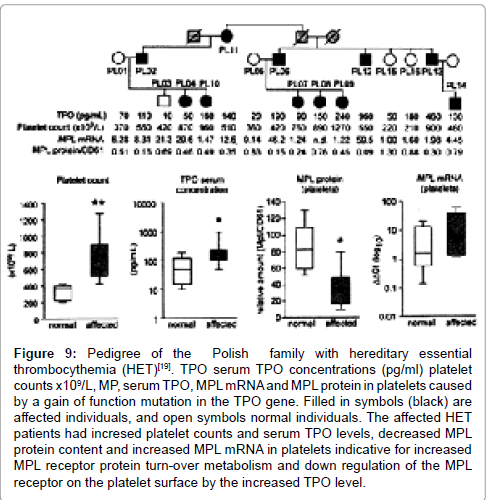
Figure 9: Pedigree of the Polish family with hereditary essential thrombocythemia (HET)[19]. TPO serum TPO concentrations (pg/ml) platelet counts x109/L, MP, serum TPO, MPL mRNA and MPL protein in platelets caused by a gain of function mutation in the TPO gene. Filled in symbols (black) are affected individuals, and open symbols normal individuals. The affected HET patients had incresed platelet counts and serum TPO levels, decreased MPL protein content and increased MPL mRNA in platelets indicative for increased MPL receptor protein turn-over metabolism and down regulation of the MPL receptor on the platelet surface by the increased TPO level.
Pathophysiology of Clinical and Bone Marrow Features in Congenital JAK2-Induced HET
There are two novel molecular variants of congenital polyclonal HET caused by a gain of function mutation in the JAK2 gene: HET caused by a gain of function mutation JAK2V617I and JAK2R564Q in the JAK2 gene [41-43]. Mead et al. described the germline mutation JAK2V617I as the sole genetic abnormality, sufficient to induce the ET phenotype of MPN in a family with autosomal dominant HET complicated by microvascular ischemic events in some of the them [41,42]. Peripheral blood and bone marrow histology are consistent with normocellular ET without features of PV [41,42]. The authors demonstrated that JAK2V617Iis the sole driver in JAK2V617I-positive individuals with typical ET peripheral blood and bone marrow features and completely normal values for haemoglobin, haematocrit, erythrocytes TPO and EPO levels [41,42]. There was a non-significant trend to increased (mean 2.4-fold) numbers of phenotypic hematopoietic stem cells (HSCs) relative to controls in the JAK2V617Ipositive persons. There were no significant differences in the numbers of other myelo-erythroid progenitor populations as determined by fluorescence-activated cell sorting, in both peripheral blood and bone marrow of the JAK2V617Ipositive ET cases. Compared with controls, however, CFU-GM were increased in the BM of JAK2V617I-positive ET cases. BFUEs were not affected, in agreement with the lack of erythroid phenotype in these patients, whereas CFU-Mks were slightly increased in the BM. After stimulation with granulocyte colony-stimulating factor (G-CSF), TPO and EPO of peripheral blood CD33+ myeloid and CD34+stem and progenitor cells, significant differences in congenital JAK2V617I and acquired JAKV617F mutated cells as compared to controls [42]. The response to G-CSF was increased in JAKV617I and more prominent in JAK2V617F mutated HSC. In signalling and transcriptional experiments assays, JAK2V617I showed more activity than wild type JAK2, but substantially less than JAK2V617F. After cytokine stimulation, JAK2VV617I resulted in markedly increased downstream signalling compared to JAK2 wild type and comparable with JAK2V617F. The responses to TPO were equally increased in JAK2V617I and JAK2V617F and the response to EPO was normal in congenital JAKV617I but increased in acquired JAK2V617F. These findings demonstrate that heterozygous congenital JAK2V617I mutation induces sufficient cytokine hyperresponsiveness of the HSC to induce a homogeneous ET phenotype in blood and bone marrow without PV features [42]. In congenital HET caused by the heterozygous JAK2V617Imutation the responses to TPO are equally increased in germline JAK2V617Iand somatic JAK2V617F mutated CD33 and CD34+ cells , but the responses to EPO are normal in germline mutated JAK2V617I and increased in acquired somatic JAK2V617Imutated hematopoietic progenitor cells (Figure 10), thereby explaining why the heterozygous JAK2V617I germline mutation is associated with ET without PV features [42].
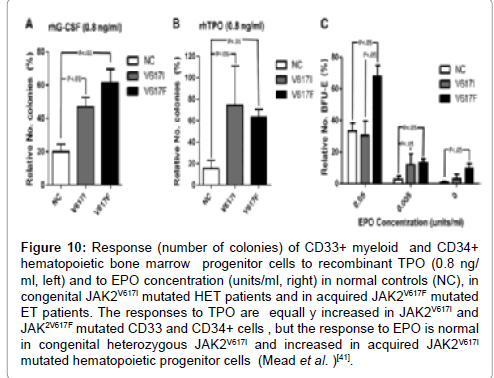
Figure 10: Response (number of colonies) of CD33+ myeloid and CD34+ hematopoietic bone marrow progenitor cells to recombinant TPO (0.8 ng/ ml, left) and to EPO concentration (units/ml, right) in normal controls (NC), in congenital JAK2V617I mutated HET patients and in acquired JAK2V617F mutated ET patients. The responses to TPO are equall y increased in JAK2V617I and JAK2V617F mutated CD33 and CD34+ cells , but the response to EPO is normal in congenital heterozygous JAK2V617I and increased in acquired JAK2V617I mutated hematopoietic progenitor cells (Mead et al. )[41].
Another novel heterozygous JAK2R564Q mutation has been identified in another family with autosomal dominant HET [43]. The growth promoting effects of JAK2R564Q were much milder than those of acquired JAK2V617F mutation. The authors found higher levels of STAT1 and STAT3 in cells expressing acquired JAK2V617F , compared to congenital JAK2R564Q. Total STAT1 levels were increased with acquired JAK2V617F and with congenital JAK2R564Q expression as compared to wild type JAK2 but this effect was more prominent with the somatic JAK2V617F mutation. Platelets were isolated from 3 members of the family with the JAK2R564Q mutation and subject to western blot analysis. Phosphorylation of JAK2 was increased in the JAK2R564Q-positive family members (R564Qand R564Q) compared with the father, who is negative for the mutation (WT). The growth characteristics of the JAK2-expressing cell lines in response to TPO treatment were then determined using 3-(4,5-dimethylthiazol-2-yl)-2,5- dimethyltetrazolium bromide assays. Cells expressing JAK2V617F , either with or without JAK2R564Q, were factor-independent, and proliferation was significantly increased from wild type JAK2-expressing cells in the absence of, and at all concentrations of, TPO. JAK2R564Q-expressing cells also showed significantly increased proliferation compared with wild type JAK2 cells, although cell proliferation was much less striking than with acquired JAK2V617F (Figure 10). An overall increase in downstream signaling in mutant JAK2R564Q cells was further demonstrated by the upregulated tyrosine-phosphorylation of proteins in germline JAK2R564Q-expressing cells as compared to wild type JAK2, and this was even more robust in the JAK2V617F-expressing somatic mutants. Similar increased signaling was observed in JAK2R564Q-positive patients by the demonstration that increased phosphorylation of JAK2 protein in platelets isolated from 3 members of the family with the JAK2R564Q mutation as compared to a JAK2 wild type family [43]. In the absence of TPO, and at all concentrations of TPO, the growth charateristics of germline JAK2R564Q-expressing cells also showed significantly increased proliferation, compared to JAK2 wild type cells, but this was much less striking than with acquired JAK2V617F thereby confirming the findings in JAKV617I HET and explaining why heterozygous JAK2V617I and JAK2R564Q are associated with ET without PV features (Figure 10). These difference do explain why acquired JAK2V617F mutated ET frequently shows features of PV with low serum EPO and increased bone marrow cellularity duet o increased erythropiseis (Figure 11) [35,37].
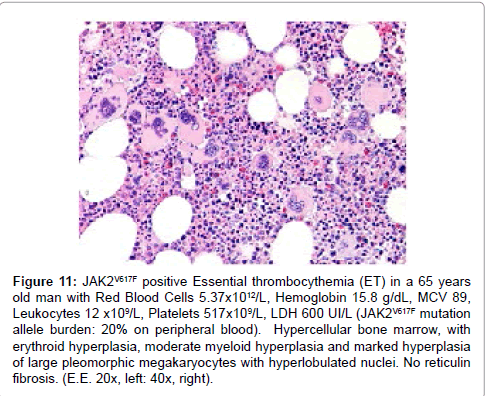
Figure 11: JAK2V617F positive Essential thrombocythemia (ET) in a 65 years old man with Red Blood Cells 5.37x1012/L, Hemoglobin 15.8 g/dL, MCV 89, Leukocytes 12 x109/L, Platelets 517x109/L, LDH 600 UI/L (JAK2V617F mutation allele burden: 20% on peripheral blood). Hypercellular bone marrow, with erythroid hyperplasia, moderate myeloid hyperplasia and marked hyperplasia of large pleomorphic megakaryocytes with hyperlobulated nuclei. No reticulin fibrosis. (E.E. 20x, left: 40x, right).
Increased plasma TPO levels produced by liver cells caused by a gain of function mutation in the TPO gene on chromosome 3q27 do not affect the hematopoetic stem cells but selectively induced proliferation and differentiation of polyclonal large megaksaryocytes and large platelets leading to the platelet-mediated arterial thrombophilia in hereditary essential thrombocythemia (HET) without features of PV. While not treated with myelosuppressive agents spontaneous evolution of normocellular HET into myelofibrosis in two patients and acute myeloid leukemia in a third case at ages around 70 years. Congenital HET caused by heterozygous gain of function mutations in the JAK2 gene is also associated with the clinical picture of ET without any feature of PV during long-term follow-up. Acquired JAK2V617F ET typically shows features of PV with low serum EPO, the presence of EEC and increased erythropioesis and peomorphic megakaryocytes in the bone marrow.