Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2016) Volume 5, Issue 4
Keywords: Inflammation; Transcription factors; Neuropathic pain; Collagen induced arthritis; Stavudine
Despite chronic pain being a widespread health issue, current management and treatments for the condition are far from satisfactory [1]. There are three key causes for chronic pain: tissue injury leading to inflammatory pain; nerve damage causing neuropathic pain; and tumour growth eliciting cancer pain [2].
Pain is one of the classic signals of the inflammatory process. Inflammatory conditions alone can lead to debilitating and persistent pain [3]. Inflammation can also be a consequence of conditions such as neuropathic pain, where inflammation is secondary to damage to the somatosensory nervous system [4]. Inflammatory mediators, such as prostaglandins, nerve growth factor, nitric oxide, cytokines, and chemokines are released from injury sites and can activate or modify nociceptor activities. This directly elicits various forms of pain, resulting in peripheral and central sensitisation and ultimately, a chronic pain state [5-7]. In fact, inflammatory reactions increase nociceptor responsiveness, allowing low-intensity stimuli access to the nociceptive pathway and produce pain [8]. In line with this, a widely used approach to treat chronic pain is by non-steroidal anti-inflammatory drugs (NSAIDs), which prevent nociceptor sensitization. NSAIDs inhibit the synthesis of prostaglandins, hence diminishing peripheral and central sensitization. However, it is not fully understood whether the inflammatory mediators cause and or maintain neuropathic pain.
Several studies suggest that the transcription factor Nuclear Factor kappa B (NFκB) could be involved in pain [9]. A significant increase in the percentages of activated NFκB immunoreactive neurons and GFAP-positive cells under inflamed conditions was reported in the dorsal root ganglia of rats [10].
In addition, recent evidence suggests the involvement of Peroxisome proliferator-activated receptors (PPARs) in pain regulation [4,11,12]. These nuclear receptors are primarily known for their roles in metabolic processes and have garnered recent interest in mediating inflammatory responses [13-15]. Pioglitazone, a PPARγ synthetic ligand, has been shown to present anti-nociceptive and anti-edematogenic effects to various animal models [16]. In animal models of neuropathic pain, the endogenous PPARγ agonist, 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2, a cyclopentanone prostaglandin), decreased mechanical and cold hypersensitivity in a dose-dependent manner [17]. Similar findings were reported using rosiglitazone, a synthetic PPARγ agonist [17]. The 15d-PGJ2 was also effective in alleviating inflammatory pain [6]. In addition, LoVerme et al. [18] investigated the effects of the PPARα agonists GW7647 and Palmitoyl Ethanilamide (PEA), and both reduced hyperalgesia in acute and inflammatory pain models.
Hence, we hypothesize that alterations in the expression of these transcription factors in the CNS may contribute to exacerbate allodynia and hyperalgesia. Therefore, it is relevant to investigate whether these transcription factors are changed during chronic pain. The aim of this study was to investigate whether the expression of NFkB and PPAR subtypes, γ and α, is affected by different models of chronic pain, including a model of inflammatory and two different models of neuropathic pain, a nerve injury model and the Anti-Retroviral (ART) model of peripheral neuropathy.
Materials and antibodies
Antibodies used were PPARγ H-100: sc-7196 (Santa Cruz), PPARα P0369 (Sigma), anti-p65 (Cell signalling), β-actin (Abcam). TNFα was obtained from Roche. All other reagents were obtained from Sigma and Invitrogen.
Ethical standards
All animal experiments conformed to the British Home Office Regulations (Animal Scientific Procedures, Act 1986) and International Association for the Study of Pain guidelines [19] for the care and use of animals. We followed the ARRIVE guidelines for reporting the behavioral studies in preparing this report [20].
Animals
In this study, three different models of pain were investigated: Collagen Induced Arthritis (CIA) mice as model of inflammatory pain [21,22]; and Spinal Nerve Transection (SNT) and Anti-Retroviral Neuropathy (ART) rats as models for neuropathic pain. Animals were housed and maintained at a temperature of 21 ± 2ºC. Sample size estimation was not conducted, as the group number was according to the need of the current study, which is mainly a molecular biology, based study. Animals were fed with normal chow food (RM1 pelleted form; Special Diet Services, Essex, UK) and tap water ad libitum, and allowed to acclimatize in their housing environment for at least 48 h after arrival.
Brain tissue from 11 mice (5 Controls; 6 CIA) was provided by Julia Inglis (Kennedy Institute). Adult male DBA/1 mice (Charles River) ages 10–12 weeks were used. Mice were immunized by subcutaneous injection at the base of the tail with 2 50-μl injections of bovine type II collagen (2 mg/mL) in CFA (Becton Dickinson, Twickenham, UK), as described previously [22]. Fourteen to 28 days following immunization the mice developed arthritis and were sacrificed [22]. The rationale for choice of route of administration and drug dose were based on your previous publication.
For the ART and the SNT models, male adult Wistar rats (180–200 g; Charles River, UK) were used. Only rats that developed hind paw mechanical hypersensitivity of at least 25% change from the baseline were included. The experimenter was blinded to the treatments received and had no knowledge of the experimental group to which an animal was randomized. The rationale for choice of a brief general anesthetic, route of administration, and drug dose were based on your previous publication.
The ART model was obtained by injecting the HIV antiretroviral drug stavudine (d4T) in Wistar rats (n=4 per group) [23]. After 2 intravenous injections, i.e. via a tail vein under a brief general anaesthesia (1–2% isoflurane [Abbott, UK] in O2 and N2O at a 1:1 ratio), of d4T (a gift from Pfizer Ltd., UK; 50 mg/kg, 4 days apart), rats developed hind paw mechanical hypersensitivity, which plateaued at 21 days after initial d4T injection, as described previously [23]. Vehicle control animals received equivalent volumes of sterile saline using the same administration protocol for d4T (n=4).
For the spinal nerve transection (SNT: 4 controls; 4 treated) Wistar rats and their controls were produced at Chelsea & Westminster Hospital as described in previous publications [24]. Surgery was performed under general isofluorane anaesthesia and aseptic surgical conditions. Perioperative analgesia (0.05 ml bupivacaine, AstraZeneca, UK) and antibiotic treatment (Enrofloxacin (“Baytril”): 0.2 ml/kg, Bayer Ltd, Dublin, Ireland) were injected subcutaneously at the start of the spinal nerve transection surgery. A 1-2 cm midline skin incision was made level with the iliac crests. The left paraspinal muscles were separated from spinous processes of L4 to S2 vertebrae using blunt dissection. Using anatomical landmarks, the L6 transverse process was identified and a small laminectomy performed, exposing L4 and L5 spinal nerve roots. The left L5 was tightly ligated (4-0 Mersilk, Ethicon) and transected 1-2 mm distal to the ligation. Transection of the L5 nerve root was confirmed post-mortem in all SNT animals. The wound was sutured and animals received intraperitoneal post-operative analgesia (20% carprofen (“Rimadyl”), 0.5 ml/kg; Pfizer, UK) 4 h postsurgery. Naive animals did not undergo any surgical procedure but were transported and housed in their cages in the surgical room for an equivalent period.
Behavioural testing
For the CIA model, animals were scored for clinical signs of inflammation, as follows: 0: Normal; 1: Slight swelling and/or erythema; 2: Pronounced edematous swelling; and 3: Joint rigidity. Each limb was graded, thus allowing a maximum score of 12 per mouse. This experiment was repeated on 3 separate occasions [22]. Thermal hyperalgesia was assessed using the Hargreaves plantar apparatus (Ugo Basile, Varese, Italy), as described previously [22]. The mice were placed in the equipment used to assess hyperalgesia on at least 2 occasions prior to pain evaluation, in order to reduce stressinduced behavioral changes. Thermal hyperalgesia was assessed using the Hargreaves plantar apparatus (Ugo Basile, Varese, Italy). Briefly, mice were placed in a Perspex box, and an increasing thermal stimulus was delivered to the plantar surface of the hind paw. The amount of time until lifting of the paw was recorded.
Behavioural data using the hind paw mechanical hyper-sensitivity are provided to confirm the validity of the models (Supplementary information, Figures S1A-S1B) [25]. Animals were tested in individual Plexiglas observation chambers (23 × 18 × 14 cm) by a single observer, ‘blinded’ to group allocation. The hind paw withdrawal threshold (PWT) in response to punctate static mechanical stimulation was assessed 14 days post-injury or 18 days following the first d4T injection using an electronic ‘von Frey’ device (Somedic AB, Hörby, Sweden). The calibrated force transducer (0.5 mm2 diameter tip) was manually applied to the mid-plantar surface of both left and right hind paw alternately (8-15 g/s) until an active limb withdrawal response was observed. The threshold value was calculated as the mean of five measurements. Two sessions of habituation (40-50 min each) to the testing area were conducted. Then, two baseline values were obtained for all rats prior to surgery. Mechanical hypersensitivity was defined as a post-operative change in the hind paw withdrawal of at least -30% from baseline for the SNT model and -25% from baseline for the ART model.
Brains from all experiments were sub-dissected into frontal cortex, midbrain, brainstem, cerebellum, and spinal cord. All tissue was snapfrozen and kept at -80°C for further analysis.
Cell culture
Mouse neuroblastoma of the N2a cell line were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin on a 35 mm diameter plates. Cells were grown in a 5% CO2 incubator at 37°C and were incubated overnight with TNFα (30 ng ml-1).
Western blot
Brain regions and cells were homogenized in radioimmunoprecipitation assay buffer (RIPA; 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl, pH 7.2) supplemented with Roche Complete protease® inhibitor cocktail. Proteins were separated on 10% sodium dodecyl sulphatepolyacrylamide (SDS-PAGE) gels and transferred to Immobilon-P polyvinylidene fluoride (PVDF). Following appropriate blocking, membranes were incubated with a primary antibody (all at 1:1000 dilution) overnight at 4°C and then incubated with secondary antibodies. Membranes were developed using ECL™ (GE Amersham, UK) reagents and Hyperfilm ECL™ audioradiography film in an automated developer (Konica, SRX 101A). The intensity of the bands was quantified by densitometry using Image J software (NIH) and normalized to β-actin.
Statistical analyses
The sample number was based on biostatistical advice and previous experience, indicating that group sizes of 4-5 animals are sufficient to detect even subtle changes for protein expression analysis. Data analysis was performed with GraphPad Prism version 5.01. Unpaired t-tests were used to compare the expression of transcription factors and pro-inflammatory mediators in control versus CIA, ART, and SNT models. In all cases, p < 0.05 was considered statistically significant.
The expression of PPARs and NFκB is altered in animal models of chronic pain
To allow better understanding towards the underlying mechanisms involved in the generation of chronic pain in the CNS, we investigated the potential changes in the expression of transcription factors NFkB and PPARs in the brain of CIA, SNT, and ART models of chronic pain. Because multiple areas all over brain are involved in pain, including those involved in sensory-discriminative, cognitive/affect responses and endogenous pain control, from cortex to brainstem, we decided to test the expression of transcription factors in frontal cortex, midbrain, brainstem and cerebellum.
Western blot analysis for PPARγ in different brain regions revealed a general reduction in the levels of these receptors in the frontal cortex, brainstem cerebellum of all the pain models (Figures 1A-1D). In contrast, we observed the opposite tendency in NFκB levels, with marked increases in p65 fragment in most brain regions of SNT model of neuropathic pain, and midbrain and brainstem of the CIA and ART models (Figure 2). On the other hand, no major changes were detected in PPARα expression in brains of the SNT and CIA models, although PPARα levels were reduced in certain brain areas of the ART model of pain (Figure 3).
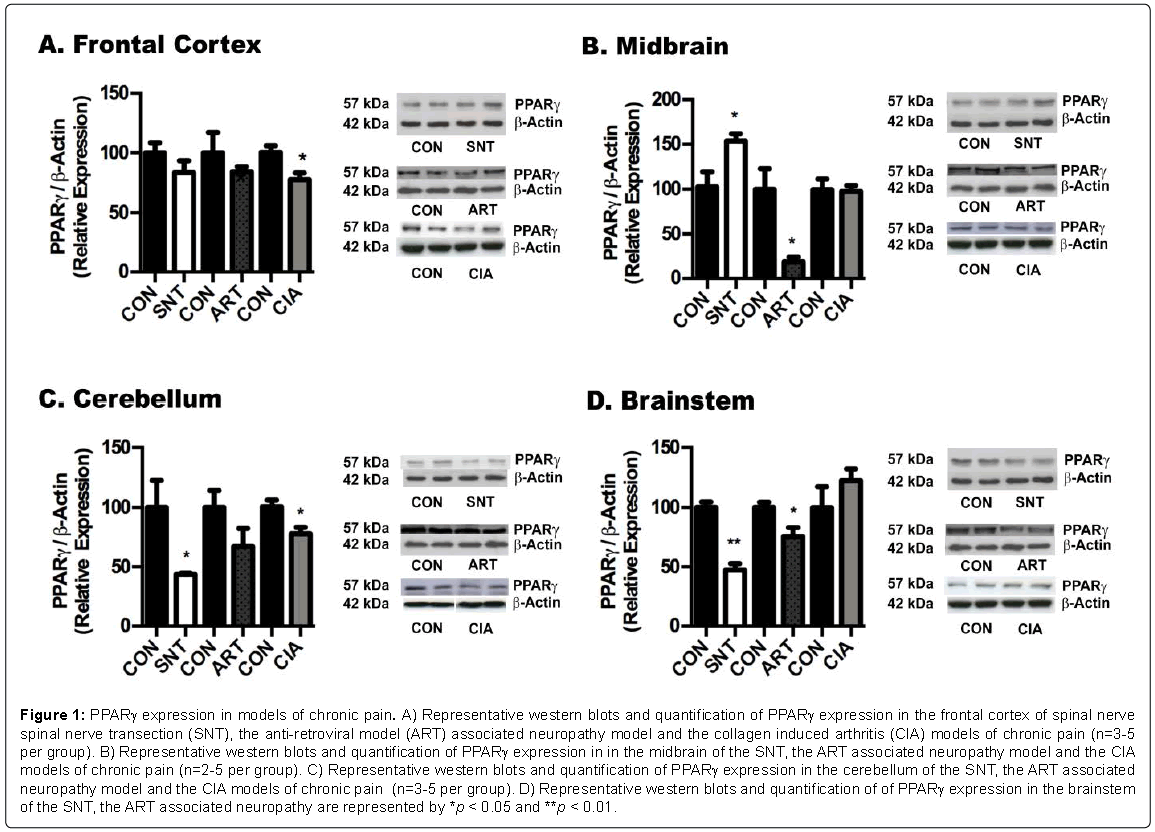
Figure 1: PPARγ expression in models of chronic pain. A) Representative western blots and quantification of PPARγ expression in the frontal cortex of spinal nerve spinal nerve transection (SNT), the anti-retroviral model (ART) associated neuropathy model and the collagen induced arthritis (CIA) models of chronic pain (n=3-5 per group). B) Representative western blots and quantification of PPARγ expression in in the midbrain of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n=2-5 per group). C) Representative western blots and quantification of PPARγ expression in the cerebellum of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n=3-5 per group). D) Representative western blots and quantification of of PPARγ expression in the brainstem of the SNT, the ART associated neuropathy are represented by *p < 0.05 and **p < 0.01.
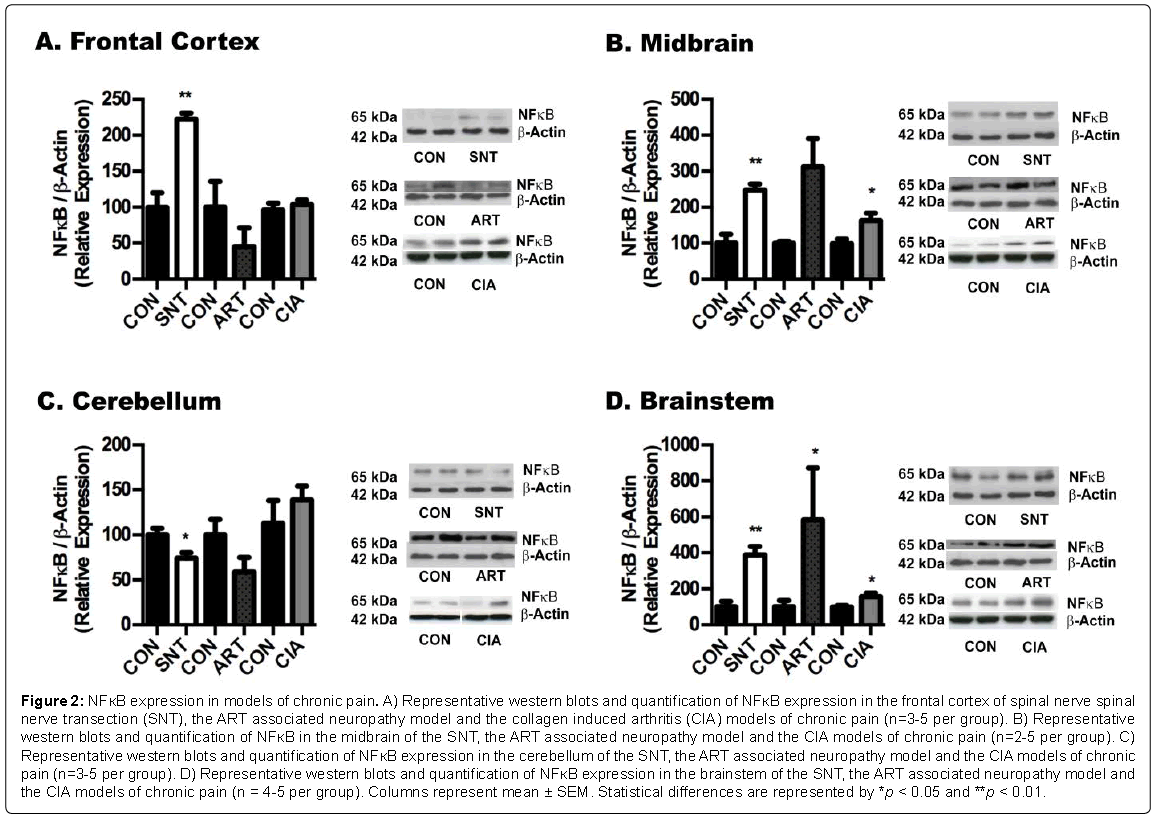
Figure 2: NFκB expression in models of chronic pain. A) Representative western blots and quantification of NFκB expression in the frontal cortex of spinal nerve spinal nerve transection (SNT), the ART associated neuropathy model and the collagen induced arthritis (CIA) models of chronic pain (n=3-5 per group). B) Representative western blots and quantification of NFκB in the midbrain of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n=2-5 per group). C) Representative western blots and quantification of NFκB expression in the cerebellum of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n=3-5 per group). D) Representative western blots and quantification of NFκB expression in the brainstem of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n = 4-5 per group). Columns represent mean ± SEM. Statistical differences are represented by *p < 0.05 and **p < 0.01.
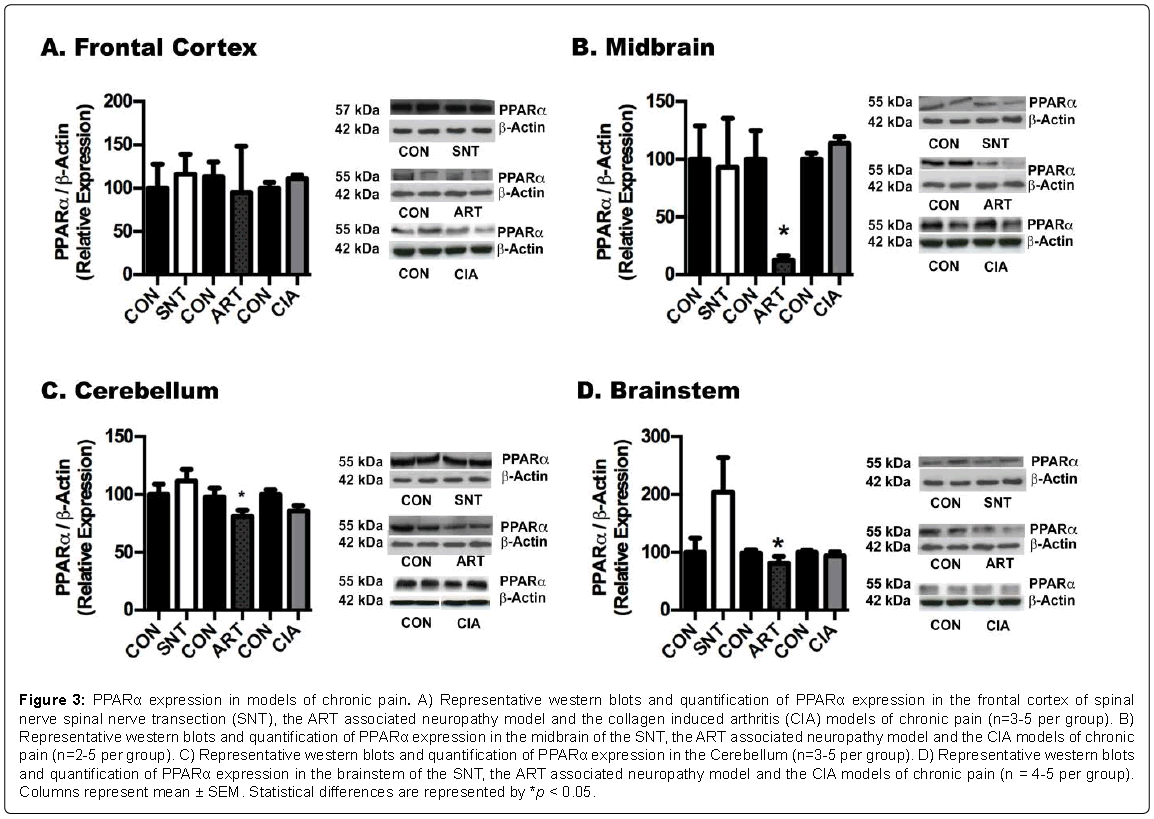
Figure 3: PPARα expression in models of chronic pain. A) Representative western blots and quantification of PPARα expression in the frontal cortex of spinal nerve spinal nerve transection (SNT), the ART associated neuropathy model and the collagen induced arthritis (CIA) models of chronic pain (n=3-5 per group). B) Representative western blots and quantification of PPARα expression in the midbrain of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n=2-5 per group). C) Representative western blots and quantification of PPARα expression in the Cerebellum (n=3-5 per group). D) Representative western blots and quantification of PPARα expression in the brainstem of the SNT, the ART associated neuropathy model and the CIA models of chronic pain (n = 4-5 per group). Columns represent mean ± SEM. Statistical differences are represented by *p < 0.05.
Effect of pro-inflammatory cytokines on the expression of PPARs and NFκB
In order to confirm whether the changes in PPARs and NFκB expression in the animal models were consequence of the inflammatory component of the model, we repeated the same measurements in N2a, which were treated with either vehicle or with 30 ng ml-1 of TNFα, because this cytokine has been found increased in CIA models [26]. When treated with TNFα, N2a cells shown a significant decrease in PPARα (c.24%, p < 0.05) and PPARγ expression (c.19%, p < 0.05) (Figures 4A and 4B respectively). On the other hand, a significant increase (c.65%, p < 0.005) in p65 was detected under inflammatory conditions (Figure 4C), suggesting that changes in inflammatory mediators may affect the transcription of PPARs and NFkB, thus explaining the changes detected in animal models of chronic pain.
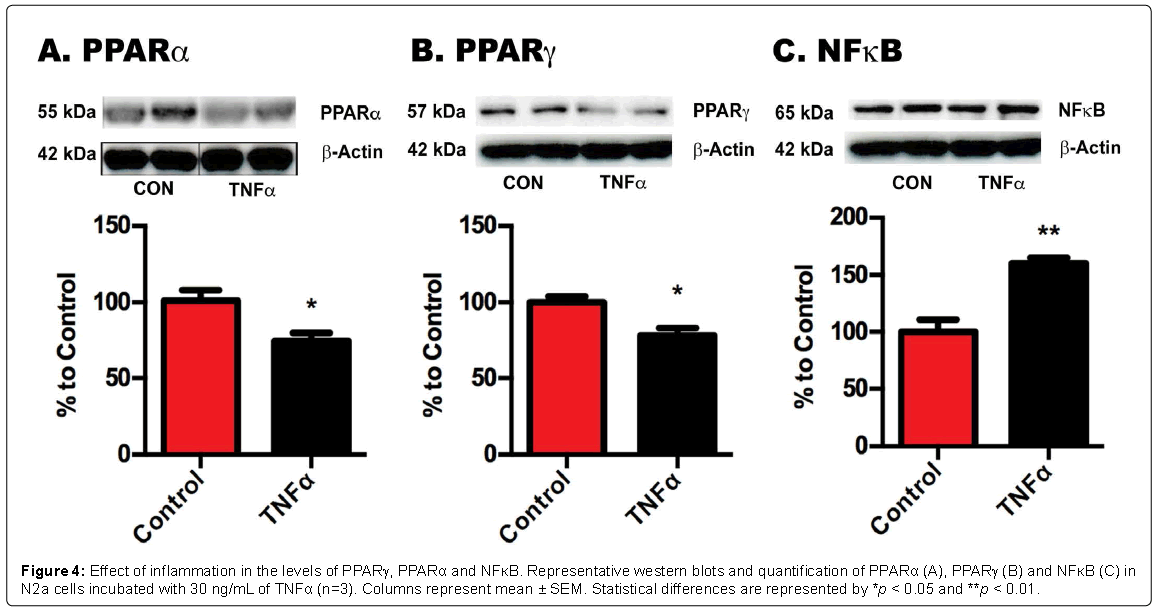
Figure 4: Effect of inflammation in the levels of PPARγ, PPARα and NFκB. Representative western blots and quantification of PPARα (A), PPARγ (B) and NFκB (C) in N2a cells incubated with 30 ng/mL of TNFα (n=3). Columns represent mean ± SEM. Statistical differences are represented by *p < 0.05 and **p < 0.01.
Our results identified substantial changes in the expression of PPARγ and NFκB across various brain regions in different models of persistent pain, in particular those associated with inflammatory pain, such as the CIA model. This was further confirmed in vitro, showing that changes in PPARα, PPARγ, and NFκB expression in cultured N2a cells under inflammatory conditions suggested that the inflammatory component of CIA models were likely to be involved in the changes of transcription factors.
So far, there are no publications reporting changes in the expression of PPARs in animal models of pain. However, it has been previously suggested that PPARs might play a role in modulating thermal and pain sensations due to their prominent expression in the thalamus, particularly in the posterior part of the ventral medial nucleus, a site responsive to pain and cold stress and on spinal cord [27,28].
Recent publications have suggested PPARγ as a new target for treating chronic pain [17,29]. Treatment with PPARγ ligands, such as pioglitazone, in the SNT model indicates that pioglitazone alleviates neuropathic pain through the attenuation of the up-regulation in proinflammatory cytokines [30]. The effect of pioglitazone in reducing pain may be mediated by a decrease in glial activation and through neuropathic non-genomic and genomic activity [31]. It has been shown that inflammatory cytokines and oxidative stress decrease the expression of PPARγ mRNA in adipocytes [32] and thiazolidinediones reverse this effect [33]. Similarly, we have previously demonstrated that certain combinations of inflammatory cytokines can decrease PPARγ gene transcription and PPRE activity in neuronal cells [33], and this effect is suppressed by incubation with NSAIDs. In line with this, it has been reported that TNFα suppresses PPARγ2 transcription by inhibiting the binding of C/EBPδ to the PPARγ2 promoter in adipocytes [35]. However, we did not observe changes in the levels of pro-inflammatory cytokines in brain areas of the SNT model and in the ART model (data do not shown). This result is in agreement with previous publications reporting neither peripheral nor central inflammatory cytokine secretion, or neuronal death, or metabolic dysregulation contributing to the development of hyperalgesia in the model of stavudine orally-induced hyperalgesia (the ART model) in rats [36]; however, results in HIV patients taking this medication seem to indicate alterations in the levels of adiponectin, which is modulated by TNFα [37]. Interestingly, PPARγ agonists have been involved in the regulation of this adipose-specific plasma protein that possesses antiatherogenic properties [38]. Performing further analysis including other inflammatory mediators such as chemokines in the brain of SNT and ART models would be necessary to address this point [39].
Both in vitro and in vivo studies have demonstrated that neuronal PPARγ prevents COX-2 increase, which in turn may reduce prostaglandin synthesis [40]. Hence, the reduction of PPARγ in pain models might contribute towards the loss of control of COX-2 upregulation, ultimately causing pain. Alternatively, the analgesic effect of PPARγ agonists could rely upon endogenous opioid activity [6]; PPARγ may modulate the transcription of pain receptors, such as opioid receptors, and consequently affect nociception. Interestingly, both natural and synthetic PPARγ agonists show rapid effects, within 5 minutes of injection, suggesting a transcription-independent pathway of action [11] and mechanism of action. The anti-allodynia effects of PPARγ agonists are PPARγ-dependent, which confirms its involvement in pain regulation [17].
Although PPARα synthetic agonists, such as fenofibrates, have shown beneficial effects in acute and chronic models of inflammatory and neuropathic pain [16,18,41,42], no major changes were observed in their expression in the CIA and SNT models of chronic pain in the present study. However, in diet-induce obese rats the levels of PPARα were found down-regulated in spinal cord, and facilitated the susceptibility to peripheral inflammatory challenge by increasing inflammatory response, contributing to augmented peripheral inflammation and inflammatory hyperalgesia [43]. This is in agreement with the reduced levels of PPARα found in the ART model and in cells incubated with TNFα. Similar to the PPARγ, it has been hypothesised that PPARα agonists decrease pain via a non-transcriptional mediated mechanism, most likely regulating the activity of ion channels [4]. PPARα has been demonstrated to affect NFkB expression [41], as the PPARα agonist Fenofibrate upregulates IκBα expression, leading to the repression of the p50 subunit of NFkB and C/EBP [44].
The increased NFκB expression in the models of chronic pain observed in the present study is in agreement with existing publications that suggest that NFκB is regulated by inflammation and could be a mediator of pain [9,45]. In rat models of paw inflammation, R-flurbiprofen has both antinociceptive and anti-inflammatory effects [46]. Other studies demonstrate that R-flurbiprofen potently inhibits NFκB activation and its target genes [45], suggesting the involvement of NFκB in both the hyperalgesic and inflammatory component of the CIA model. On the other hand, activated p50 would seem to be a very interesting target to investigate in pain since p50 knockouts mice demonstrated reduced acute and inflammatory nociceptive responses [47].
In summary, PPARγ and NFκB seem to be oppositely regulated in models of chronic pain. In fact, PPARγ has been shown to be able to block NFκB actions by several mechanisms, including I-κBα induction [48] or by transcriptional transrepression, also known as squelching, receptor mutual antagonism, or cross-coupling, therefore interfering with NFκB transcriptional activity [49,50].
In conclusion, the results of this study identify alterations in a common signalling pathway in different models of persistent pain and suggest that changes in PPARs and NFκB seem to be secondary to neuroinflammation, therefore contributing to further exacerbation of pain symptomatology. The result of interferences with the PPAR signalling pathway that has become evident in pain has far reaching consequences that extend beyond the pain scenario, affecting other conditions where PPARs have a role, including neurological pathologies such as Alzheimers disease [34]. Further research needs to be highlighted in these areas, as there is the potential that modification of this signalling pathway can ameliorate or even reverse these pathologies.
Please, see ethical section in Materials and Methods.
This project was partly funded by a grant from the Fundacio Marato TV3, ref 072610 (Catalonia, Spain). We thank Prof. Andrew S. Rice for his collaboration in the ART and SNT models.