Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2014) Volume 2, Issue 4
Adult T-cell leukemia/lymphoma (ATL) is a highly aggressive leukemia/lymphoma. The long clinical latency and low incidence of ATL indicate that ATL is an age-related disease with genetic changes that are involved in malignant transformation and monoclonal expansion of human T lymphotropic virus type 1 (HTLV-1) infected cells. The question remains as to how the ATL cells acquire monoclonal proliferation rather than undergo senescence. It was implied that aberrant cellular proteins such as p53, JAK-STAT and Bcl-xL were associated with the development of ATL. HTLV-1-infected cells and ATL cells (partial) express CD30. Soluble CD30 (sCD30) may impair the interaction of CD30 ligand+ cells with CD30+ ATL cells. Probably HTLV-1-infected T-cells migrate to the lung due to agerelated changes of pulmonary immune system. Thereafter monoclonal proliferation of infected cells following antigen presentation forms a local network between cytokines and cytokine receptors. During the remodeling of the pulmonary tissue, CD30+ ATL cells and matrix metalloproteinases can play an important role in forming tumor microenvironment. Furthermore, soluble cytokine receptors may support the clonal proliferation and invasion of ATL cells and tumor microenvironment is created in extra nodal lesions such as lung. Accordingly, sCD30 level could be applied to detection of microenvironment formation for tumor stem cells and their monitoring in ATL.
Keywords: Adult T-cell Leukemia/Lymphoma (ATL); Human T Lymphotropic virus type 1 (HTLV-1);Soluble CD30 (sCD30); Microenvironment; Senescence
Adult T-cell leukemia/lymphoma (ATL) is a highly aggressive leukemia/lymphoma which was first proposed as a new disease entity in 1977 [1,2]. There is a long clinical latency over 50 years and low incidence of ATL from human T lymphotropic virus type 1 (HTLV-1) carriers and a thousand of ATL patients die in Japan every year [3]. However, the mechanism of ATL development remains unclear. It is indicated that ATL is an age-related disease involving genetic changes are prior to malignant transformation and monoclonal expansion of HTLV-1 infected cells. HTLV-1 exhibits several unique properties among known acute or chronic transforming retroviruses. HTLV-1 mediated T-cell transformation presumably arises from a multistep oncogenic process in which the virus induces chronic T-cell proliferation with accumulation of genetic defects followed by deregulation of cell growth [4,5]. Monoclonal proliferation of HTLV-1 infected cells is observed in who ultimately develop ATL, constituting the high risk group for ATL development [6]. The question remains as to how the ATL cells acquire monoclonal proliferation rather than undergo senescence.
It was implied that aberrant cellular proteins such as p53, signal transducers and activators of transcription (STAT) and Bcl-xL were associated with the development of ATL (Figure 1). The check point tumor suppressor p53 is mutated or detected in approximately ~40% of all ATL patients [7-10]. Though p53 mutants cannot bind with mouse double minute 2 are stabilized, even in the absence of genetic mutation or deletion, the function of p53 is or may be impaired in ATL cells, suggesting inhibition of senescence in vivo [11]. In addition, ATL cells constitutively activated the Janus kinase (JAK)-STAT pathway important for HTLV-1-induced immortalization and transformation mechanisms [12]. In physiologic conditions, activation of the JAK-STAT pathway is triggered by cytokines through cell surface receptors. Nevertheless, we observed JAK-STAT pathway activation in ATL cells without cytokine stimulation. HTLV-1 interferes with key steps of the apoptosis pathway, inducing ligand-independent transformation of human CD4+ T-cells, including constitutive phosphorylation of JAK and STAT proteins such as Stat3 and Stat5. Furthermore, the constitutive activation of the JAK-STAT in ATL cells may up-regulate Bcl-xL. The Bcl-xL expression was markedly increased in uncultured leukemic ATL cells with expression correlating with the disease severity [13]. Aberrant expression of Bcl-xL may increase ATL cells survival contributing to chemotherapy resistance. HTLV-1 infection eventually transforms cell to ATL cells by a long process involving inhibition of senescence and apoptosis with immune alteration. However, the reason why JAK-STAT pathway is consistently activated in vivo remains unclear.
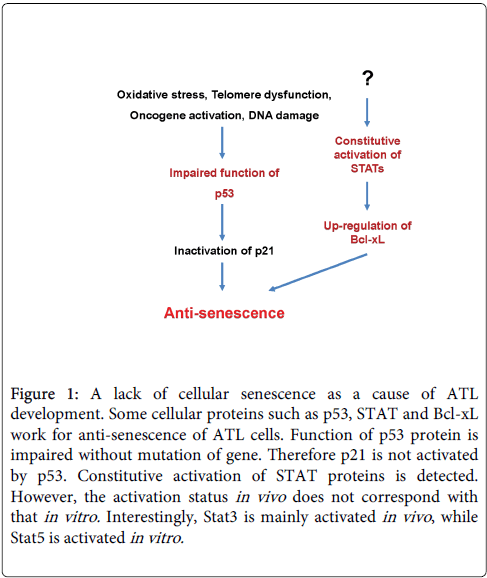
Figure 1: A lack of cellular senescence as a cause of ATL development. Some cellular proteins such as p53, STAT and Bcl-xL work for anti-senescence of ATL cells. Function of p53 protein is impaired without mutation of gene. Therefore p21 is not activated by p53. Constitutive activation of STAT proteins is detected. However, the activation status in vivo does not correspond with that in vitro. Interestingly, Stat3 is mainly activated in vivo , while Stat5 is activated in vitro.
CD30, a 120 kDa type I cell surface glycoprotein, is an unusual member of the tumor necrosis factor receptor superfamily with a duplicated segment within its extracellular domain that functions as an additional ligand binding site [14]. CD30 expression is dependent on mitogen or viral activation and proliferation of B and T-cells. Th2 clones express mRNA and membrane and soluble form of CD30 (sCD30) [15]. Tumor cells from Hodgkin lymphoma (HD) and anaplastic large cell lymphoma (ALCL) express CD30 [16]. HTLV-1 infected cells and ATL cells (partial) also exhibit CD30 and cDNAs encoding the CD30 protein cloned from expression libraries of HTLV-1+ human T-cell line HUT-102 [17,18]. Matrix metalloproteinase (MMP) cleaves CD30 with the juxta-membrane region and sCD30 is released as an extracellular region of an 85/90 kDa protein (Table 1) [19]. Interestingly, in antibody-based immunotherapy, antibody-induced CD30 shedding is catalyzed by ADAM10 but not by ADAM17 [20]. In contrast, proteasome inhibitor-induced CD30 shedding is catalyzed by ADAM17 [21]. CD30+ cells release sCD30 in vitro and in vivo, detected at low levels in the sera of healthy donors [22].
| Tumor virus | Lung legion |
| Human T lymphotropic virus type 1 (HTLV-1) | HTLV-1 associated bronchiolo-alveolar disorder (HABA) |
| HTLV-1 associated bronchopneumopathy (HAB) | |
| Adult T-cell leukemia/lymphoma (ATL or ATLL) | |
| Epstein-Barr virus (EBV) | Pyothorax-associated lymphoma (PAL) |
| Interstitial lung diseases | |
| Human herpesvirus type 8 (HHV-8) | Primary effusion lymphoma (PEL) |
Table 1: CD30 associated tumor viruses and lung legions
CD30, regulated by HTLV-1, displays activation of tumor cells dependent on cell type [23-25]. CD30 signaling induces cell proliferation in HD cells of B cell origin, whereas it promotes apoptosis in ALCL cells of T cell origin. The binding of sCD30 prevents the interaction between CD30 and its ligand which in the case of tumor cell, blocks the biological function of CD30 signaling affecting apoptosis or proliferation depending on tumor cells type [26]. Therefor sCD30 may be a biomarker and specific prognostic indicator of the risk for a lower complete response rate and relapse-free expectancy for ATL as well as other lymphoma patients including those with HD and ALCL [27,28]. Actually levels of sCD30 and soluble interleukin-2 receptor a chain are elevated in HTLV-1 carriers, becoming highly elevated in ATL [29,30]. Furthermore, sCD30 elevation is followed by acute crisis from indolent type of ATL. Accordingly, sCD30 may impair the interaction of CD30 ligand+ cells with CD30+ ATL cells. We hypothesize that CD30 signaling may induce apoptosis or cell cycle arrest of HTLV-1 infected cells during the HTLV-1 carrier phase and indolent phase of ATL (Figure 2). It is speculated that HTLV-1 mediates sCD30 production and CD30 signaling pathway is interfered by binding of sCD30 to the CD30 ligand. This process promotes survival of ATL cells, enables their monoclonal proliferation and inhibits apoptosis.
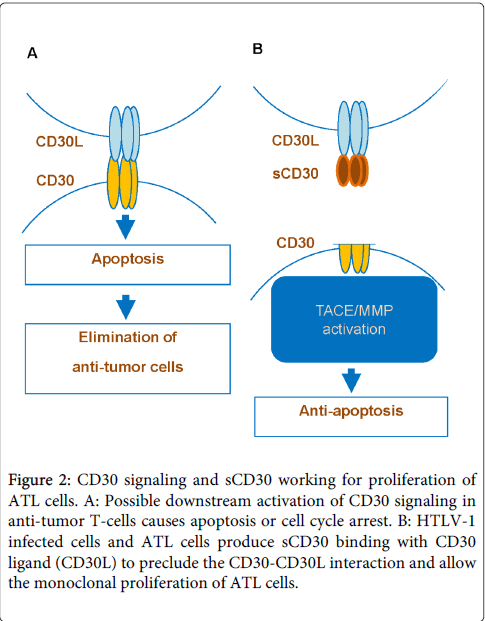
Figure 2: CD30 signaling and sCD30 working for proliferation of ATL cells. A: Possible downstream activation of CD30 signaling in anti-tumor T-cells causes apoptosis or cell cycle arrest. B: HTLV-1 infected cells and ATL cells produce sCD30 binding with CD30 ligand (CD30L) to preclude the CD30-CD30L interaction and allow the monoclonal proliferation of ATL cells.
The lungs are the preferential site for HTLV-1 infected cells [31,32]. The chest CT scans revealed abnormalities in 98 (30.1%) of 320 HTLV-1 carriers [33], and 13 (44.8%) of 29 ATL patients had leukemic infiltration [34], suggesting that this peculiar tropism is responsible for the high incidence of pulmonary involvement. From the characteristics of the feature on the lungs, they makes breathing constantly touching atmosphere. That defense against infection and various toxic substances is required, and, for this reason, the immune and metabolic functions have developed. In addition, general circulation must pass the pulmonary system. Therefore the reactions in lung affect whole-body reaction to cause immune response. Probably HTLV-1 infected T-cells migrate to the lung due to age-related changes of pulmonary immune system. Those cells express virus and induce a strong immune reaction once they invade the lung tissue [35]. Thereafter monoclonal proliferation of infected cells following antigen presentation forms a local network between cytokines and cytokine receptors. It is progressing to bronchitis and further smoldering type ATL.
Some tumor viruses such as HTLV-1, Epstein-Barr virus (EBV), and hepatitis virus type B and C induce CD30 expression on the surface of their infected cells. Especially, HTLV-1, EBV and human herpesvirus type 8 (HHV-8) are associated with lung lesions as shown in Table 2 [36,37]. For invasion of such tumor virus-infected cells into lung tissue, cleavage of collagen and elastin which are composed of lung requires the activation of MMPs. The MMPs works as an enzyme for the tissue destruction and cell infiltration as well as shedding of the cell surface cytokine and cytokine receptor. Because CD30 is one of such cytokine receptors, the soluble form enters systemic circulation and the level of sCD30 is elevated in blood. For example, serum sCD30 levels are elevated in other lung diseases such as bronchial asthma and pulmonary tuberculosis in which MMPs are activated [38,39]. Therefore, during the remodeling of the pulmonary tissue, CD30+ATL cells and MMPs can play an important role in forming tumor microenvironment.
| sCD30 | sIL-2R | |
| Production | Activation of ADAM10 and ADAM17 | Activation of MMP9 |
| ATLL cells (part) | ATLL cells (most) | |
| Activated T cells | Activated T cells | |
| Activated B cells | Regulatory T cells | |
| Viral infected cells | ||
| Dendritic cells | ||
| NK cells | ||
| Regulatory T cells (part) | ||
| Binding | CD30 ligand (CD153) | Forming IL-2 and IL-2R complex |
| ATLL cells | Regulatory T cells | |
| Monocytes / Macrophages | ||
| Neutrophils | ||
| T cells | ||
| B cells | ||
| Function | 1. Block of CD30―CD30 ligand interaction | 1. Activation of regulatory T cells |
| 2. Reverse signal to CD30 ligand (Activation) |
Table 2: Differences between soluble CD30 and soluble IL-2 receptor α chain
It is difficult to explain why it takes long time more than 50 years from HTLV-1 carriers to ATL development and why most of HTLV-1 carriers keep their carrier state for their lives without the onset of ATL. Those evidences and our experience suggest that HTLV-1 is not sufficient to transform the infected cells and clonal proliferation of HTLV-1 infected cells requires additional supports (Figure 3). The phenotype of ATL cells is mature CD4+CD25+ T-cells. They can proliferate following antigen presentation by dendritic cell [40]. Probably airborne infections activate HTLV-1 infected T-cells in lung. However, activation-induced cell death (AICD) controls the number of clonal proliferating T-cells. During senescence, aging of immune system might be a major cause of chronic infection and immunoreaction. HTLV-1 infected T-cells produce cytokines and soluble cytokine receptors. In addition, aberrant cellular proteins including p53 and STATs result in the evasion of apoptosis of activated HTLV-1 infected T-cells. Furthermore, soluble cytokine receptors may support the clonal proliferation and invasion of ATL cells and tumor microenvironment is created in extra nodal lesions such as lung (Table 2).
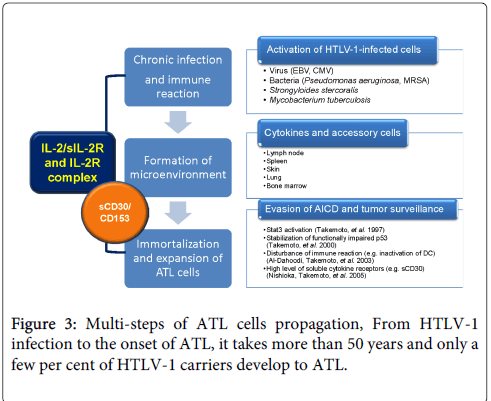
Figure 3: Multi-steps of ATL cells propagation, From HTLV-1 infection to the onset of ATL, it takes more than 50 years and only a few per cent of HTLV-1 carriers develop to ATL.
We reviewed how ATL cells monoclonal proliferation and the key factors underlying ATL cell survival, senescence, or apoptosis. These findings may lead to strategies aimed at early-stage detection and inhibition of proliferation or induction of cell death. The serum level of sCD30 could be applied to the detection of microenvironment formation for tumor stem cells and their monitoring.
S. Takemoto received research funding from Kyowa Medex Co., Ltd. (Tokyo, Japan) and Kyowa Hakko Kirin (Tokyo, Japan). These funding sources were involved in a part of data collection, but not in any study design, data analysis, data interpretation, or writing of the report. R. Pornkuna declares no competing financial interests.