Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Research Article - (2014) Volume 2, Issue 6
Somatic mutations in the JAK2, MPL and calreticulin (CALR) genes are the driver causes of clonal myeloproliferative neoplasms (MPN). Applying the WHO Clinical, Molecular and Pathologic (WHO-CMP) classification of MPN, the JAK2V617F positive ET patients comprise three phenotypes of ET: normocellular ET, hypercellular ET due to increased erythropoiesis (prodromal PV) and ET with hypercellular megakaryocytic-granulocytic myeloproliferation (ET.MGM or masked PV). The percentage of JAK2V617F mutation load is low and stable in heterozygous normocellular ET and increasingly high in hetero/homozygous PV and masked PV. The JAK2V617F allele burden is related to MPN disease burden in terms of splenomegaly, constitutional symptoms and myelofibrosis. Five distinct clonal MPNs can be distinguished: JAK2V617F mutated ET and PV; JAK2 exon 12 PV and the JAK2 wild type ET and MF caused by the somatic mutations MPL515 or CALR. JAK2 mutated trilinear MPN reflects a broad spectrum of ET, prodromal or masked PV and classical PV, but the JAK2 wild type MPL or CALR positive ET and MF lack features of PV at diagnosis and during follow-up. Bone marrow features in JAK2V617F mutated ET and PV are similar and featured by medium sized to large (pleomorphic) megakaryocytes with only a few giant forms. Bone marrow histology in MPL515 mutated ET and MF is featured by clustered small and giant megakaryocytes with hyperlobulated stag-horn-like nuclei, in a normocellular bone marrow with no features of PV. Bone marrow histology in CALR mutated ET and MF is featured by dense clustered large immature dysmorphic megakaryocytes and bulky (cloude-like) hyperchromatic nuclei similar as described in primary megakaryocytic granylocytic myeloproliferation (PMGM), which are never seen in JAK2V617F, JAK2 exon 12 and MPL515 mutated MPN.
Keywords: Myeloproliferative neoplasms; Essential thrombocythemia; Polycythemia vera; Primary megakaryocytic granulocytic myeloproliferation; Myelofibrosis; JAK2V617F mutation; MPL515 mutation; Calreticulin; CALR mutation; Bone marrow pathology
Polycythemia vera (PV) are described by Vaquez [1], Osler [2,3] and essential thrombocythemia (ET) and PV has been delineated as distinct clinical disease entities when the 1980 Rotterdam Clinical and Pathological (RCP) criteria are applied (Table 1) [4,5]. Osler’s description in 1903 of PV is charaterized by chronic cyanosis, erythrocythemia, and moderate enlargment of the spleen [2]. Osler interpreted in 1908 red painfull ‘neuralgias’in the extremities as erythromelalgia [3]. Michiels discovered between 1975 and 1980 that aspirin responsive erythromelalgia is caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia of patients with ET and PV [4,5]. Park-Weber and Watson [6] recognized that chronic polycythemia with enlarged spleen was a disease of the bone marrow. The oldest treatment of PV is phlebotomy mentioned by Osler in 1903, and 1908 [4,5]. The relationship to PV was described In 1923 Minot and Buckman described the evolution of myelofibrosis and myeloid metaplasia in 3 PV patients, who developed anemia and splenomegaly after 5-20 years follow-up [7]. The original diagnostic criteria and treatment of PV of Dameshek in 1940 and 1950 has been completely overlooked by all leading PVSG and WHO MPN investigators in the USA [8-13]. Dameshek and Henthell described between 1928 and 1937 twenty PV cases and proposed a definite set of diagnosistic criteria for PV by the presence of plethoric appearance, splenomegaly, definitely elevated erythrocyte count (>6 x 1012/L), elevated platelet count, elevated hematocrit and a hypercellular bone marrow morphology with increased erythro-megakaryo-grabulocytosis in smears of aspirated bone marrow from the iliac crest or sternum [9].
| A. The 1980 RCP major (A) and confirmative (B) criteria for prefibrotic ET A1 Persistent platelet count in excess of 400 x 109/L [4,5]. A2 Increase and clustering of enlarged megakaryocytes in bone marrow biopsy. A3 No or slight increase of reticulin fibers (RF 0 or RF 1) B1 Presence of large platelets in a peripheral blood smear B2 Absence of any underlying disease for reactive thrombocytosis and normal ESR. B3 No splenomegaly (<12 cm) or slight splenomegaly on palpation or scan (<15 cm) B4 Increase of LAP-score and no signs of fever or inflammation Exclusion criterion Ph+ chromosome and any other cytogenetic abnormality in blood or bone marrow nucleated cells |
| B. The 1980 RCP major (A) and minor (B) criteria for prefibrotic PV A1 Increased erythrocyte count above 6 x 1012/L: Dameshek 1940 [9]. A2 Raised red cell mass. Male >36 ml/kg, female >32 ml/kg: PVSG 1971-1975 [12,13] A3 Increase in bone marrow biopsy of clustered, large pleomorphic megakaryocytes with hyperlobulated nuclei and increased cellularity due to increased megakaryopoiesis erythropoiesis or typically trilinear mega-erythro-granulopoiesis. typical PV bone marrow excludes erythrocytosis. B1 Thrombocythemia, persistant increase of platelet >400 x 109/L [4,5] B2 Leukocytosis, leucocyte count >109/L and low erythrocyte sedimentation rate (ESR) B3 Raised leukocyte alkaline phosphatase (LAP) score >100, absence of fever or infection B4 Splenomegaly on palpation or on isotope/ultrasound scanning A1 or A2 plus A3 and none of B establishes erythrocythemic PV A1 or A2 plus A3 plus one of B establishes PV and excludes erythrocytosis |
Table 1: The Rotterdam Clinical and Pathological (RCP) criteria for Essential Thrombocythemia (ET) and Polycythemia Vera (PV) 1975-1980.
1975 PVSG versus 1980 RCP criteria for ET and PV
The 1975 PVSG clinical criteria used a minimal platelet count of 1000 x 109/L for the diagnosis of ET [10]. The 1975 PVSG diagnostic criteria of PV did not use bone marrow histology and excluded by definition the erythrocythemic stage 1 PV (idiopathic erythrocythemia: IE) with normal platelets, leukocytes and spleen size [11-13]. The 1980 RCP criteria of ET and PV were determined between 1975 and 1980 by careful prospective documentation of peripheral blood and bone marrow smears and bone marrow histology in the late 1970s (Table 1) [5,14]. The PVSG reduced in 1986 the minimum platelet count for the diagnosis of ET from 1000 x 109/L to 600 x 109/L [15] as the consequence of two evidence-based studies by Michiels et al. [5] and Van De Pette et al. [16]. Lengfelder et al. [17] demonstrated in 1998 that PVSG defined ET at platelet count above 600 x 10 /L overlooks 30% of early stage ET as compared to the cut-off level of 400 x 109/L (Table 1). Platelets in excess of 400 x 109/L, and increase of clustered larged megakaryocytes in a bone marrow biopsy material were found to be diagnostic for ET and excluded reactive thrombocytosis [5,17]. Between 1975 and 1980, we routinely used bone marrow histopathology and erythrocyte count above 6 x 1012/L according to Dameshek in 1940 [9] as specific clues to the diagnosis of PV to clearly differentiate PV from all variant of primary and secondary erythrocytosis (Table 1). The RCP modifications of PVSG criteria for PV include 4 changes (Table 1). The major criterion O2-saturation of >92% is deleted and replaced by bone biopsy as a major criterion (A3) to differentiate between PV and secondary erythrocytosis. Splenomegaly is used as a minor criterion (Table 1). Raised B12 (>900 ng/L) or raised B12 binding capacity (>2200 ng/L) is skipped as completely irrelevant for the diagnosis of PV (Table 1). Bone marrow histology has a specificity and sensitivity near to 100% to differentiate between the MPDs ET and PV from reactive thrombocytosis and primary or secondary erythrocytoses [14,17]. Idiopathic erythrocythemia (IE) is featured by increased red cell mass, normal spleen size, normal leukocyte and platelet counts and no clinical or laboratory evidence of primary or secondary erythrocytosis and a typical PV bone marrow histology (Table 1). The PV experts in the UK and France did not use bone marrow biopsy for the diagnostic differentiation between PV and primary or secondary erythrocytosis and therefore overlooked stage 1 erythremic PV by definition [18,19]. IE represent a significant number of early stage erythremic PV of about 10% to 15% at time of PV presentation [18,19]. The diagnostic difficulties regarding the original PVSG criteria without the use of bone marrow pathology were solved by Tom Pearson by applying on top of PVSG criteria low serum erythropoietine (EPO) levels and spontaneous endogenous erythrocyte colony formation (EEC) as specific clues to PV [20]. About half of ET patients are ECC positive and have decreased or low serum EPO levels and are in fact prodromal phases (forme frusta) of PV. Standardized and easy-to-perform commercial serum EPO assays are used for the differential diagnosis of either erythrocytosis or PV [21,22]. In a multicenter study on 241 patients, Mossuz et al. identified two thresholds of serum EPO levels, allowing a specific and correct diagnosis in 65.6% (65 out of 99 PV patients) of PVSG-defined PV patients with serum EPO levels below 1.4 U/L and in 19.7% (13 of 66 SE patients) of secondary erytrocytosis (SE) with serum EPO levels above 13.7 U/L) [21]. Consequently, about 50% of patients with increased RCM could not be diagnosed as PV or erythrocytosis indicating the needto peform a bone marrow biopsy to distinguish PV from SE (Table 1) [5,17].
Bone Classification and WHO criteria for ET, PV and PMGM
Georgii et al. discovered in 1980 prefibrotic chronic megakaryocytic gramulocytic myeloproiferation (CMGM) as the third distinct entity of primary MPD in the absence of reticulin or collagen fibrosis in bone marrow biopsy material [23]. In 1987 Michiels et al. defined strict morphological, biochemical, and cytogenetic criteria for BCR/ ABL-positive ET and chronic myeloid leukemia (CML) as a separate malignant and individual entity, whereas ET, PV and CMGM form a chronic proliferation of three hematopoietic cell lines [24]. Michiels et al. [24] and Georgii et al. (Hannover Bone Marrow Classification of CML and MPD, Table 1) [25,26] separated the Ph-positive or BCR/AB -positive CML and ET from the Ph- or BCR/ABL-negative MPDs ET, PV and CMGM based on distinct bone marrow histology findings for each of the three MPDs ET, PV and CMGM [23-28]. The difference in size and morphology of small monolobulated megakaryocytes in Phpositive CML and ET from the large pleomorphic megakaryocytes in the Ph-negative MPDs ET and PV is so obvious that cytologists and pathologists can easily distinguish [24,25]. The 1990 Hannover Bone Marrow Classification distinguished three primary prefibrotic MPDs ET, PV and chronic megakaryocytic granulocytic myeloproliferation (CMGM) from advanced fibrotic stages of MPD (Table 2) [25,26]. As myelofibrosis (MF) is a secondary event in all variants of MPD and the terms chronic idiopathic myelofibrosis (CIMF) and primary myelofibrosis (PMF) are a misconception, Georgii consequently replaced the terms CIMF and PMF by CMGM and used grading of reticulin fibrosis (RF) and increase of reticulin and collagen myelofibrosis (MF) for staging of prefibrotic, early fibrotic and overt and advanced fibrotic MPDs ET, PV and CMGM [8,9]. Prefibrotic CMGM is the third MPD entity without features of ET, PV or CML and its diagnosis is based on the presence of loose to dense clustering of large megakaryocytes with immature cytoplasm and cloud-like nuclei not seen in ET, PV and CML [25-28]. The term CMGM of the Hannover Bone Marrow Classification has illogically replaced again by Thiele and Vardiman by chronic idiopathic myelofibrosis (CIMF) in the 2001 WHO classification [29], and as PMF by Tefferi and Thiele in the 2008 WHO classification (Figure 1) [30]. The diagnosis of prefibrotic CMGM is based on the association of hypercellular ET with the presence of large immature megakaryocytes with immature cytoplasm and cloud-like nuclei not seen in ET and PV (Table 2) [31]. The WHO Clinical, Molecular and Pathological (2014-CMP) classification of Michiels et al. of the myeloproliferative neoplasms extended the CMGM concept of Georgii et al. and replaced the term CMGM by primary megakaryocytic granulocytic myeloproliferation (PMGM, Figure 1) [32,33].

Table 2: The Dameshek one cause hypothesis of trilinear PV [33] in 1950 and Vainchenker’s discovery of heterozygous and homozygous JAK2V617F somatic mutations [36] in 2005 as the driver cause of trilinear MPN with clinical and pathological manifestations of Essential Thrombocythemia (ET), Polycythemia Vera (PV) and ‘Primary’ Megakaryocytc Granulocytic Myeloproliferation (PMGM) and secondary Myelofibrosis (MF) [27,31] Designed by Michiels in 2005 [31].
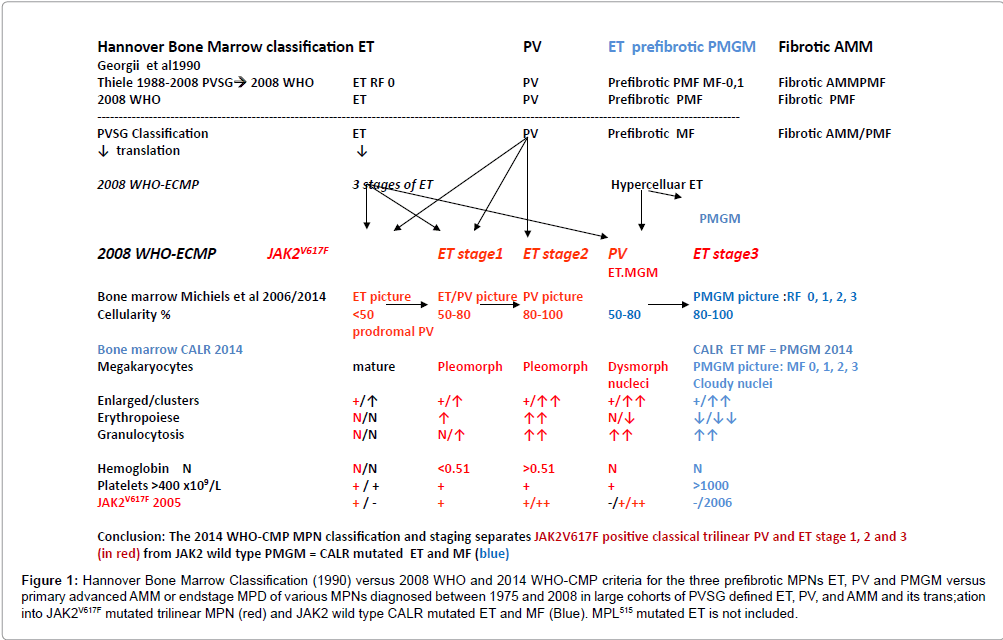
Figure 1: Hannover Bone Marrow Classification (1990) versus 2008 WHO and 2014 WHO-CMP criteria for the three prefibrotic MPNs ET, PV and PMGM versus primary advanced AMM or endstage MPD of various MPNs diagnosed between 1975 and 2008 in large cohorts of PVSG defined ET, PV, and AMM and its trans;ation into JAK2V617F mutated trilinear MPN (red) and JAK2 wild type CALR mutated ET and MF (Blue). MPL515 mutated ET is not included.
JAK2V617F mutated trilinear MPNs in ET and PV: Dameshek- Vainchenker’s Disease
In 1950, Dameshek (1900-1969) proposed two highly speculative possibilities as the cause of trilinear PV (erythrocythemia, thrombocythemia, granulocythemia: either excessive bone marrow stimulation by an unknown factor, or the lack or diminution of an inhibitory factor [34,35]. This original observation of PV as a trilinear MPD has been proven to be correct by Vainchenker’s discovery in 2005 of the somatic JAK2V617F mutation as the driver cause of he trilinear MPNs ET, PV and MF [36]. This has been rapidly confirmed by Green UK, Kralovics Europe and Levine USA [37-39]. On position 617 of the JAK2 JH2 domain Valine (V) is replaced by Fenylalanine (F) in the JAK2V617F mutation and induces a loss of inhibitory activity of the JH2 pseudokinase part on the JH1 kinase part of JAK2, leading to enhanced activity of the normal JH1 kinase activity of JAK2[36]. The JAK2V617F makes the mutated hematopoietic stem cells hypersensitive to hematopoietic growth factors TPO EPO, IGF1, SCF and GCSF, resulting in PV as a trilinear MPN (Table 2) [31]. Detection of JAK2V617F has become the first intention diagnostic test for ET and PV (Tables 3 and 4) [31]. The prevalence of the JAK2 V617F mutation in PVSG defined PV is 95% and about 50% in ET and MF [31]. The JAK2V617F mutation load in gramulocytes is usually low in heterozygous ET, less that 10 to maximal 50% and either low with less than 50% (heterozygous homozygous) or high between 50 to 100% (homozygous) in PV [40,41]. Patients with hypercellular ET, masked PV and PV homozygous for the JAK2V617F mutation patients are at high risk for myeloid metaplasia of the spleen with splenomegaly and bone marrow transformation into myelofibrosis (MF) [42]. The 2005 concept according to Vainchenker and Michiels is that heterozygous JAK2V617F mutation leading to constitutively activated megakaryocytes with increased sensitivity to TPO and EPO is enough to induce ET with the production of constitutively activated (hypersensitive) platelets (Table 2) [31]. So-called heterozygous PV with allele load less than 50% appeared to be hetero/homozygous for the JAK2V617F mutation at the EEC level in blood and bone marrow for the JAK2V617F mutation, whereas ET patients are heterozygous for the JAK2V617F mutation at the EEC level with a maximal JAK2V617F mutation load ranging from low to maximal 50% [43,44]. The change from heterozygous ET into homozygous masked or overt PV is due to the loss of 9p heterogeneity (9P LOH) of the JAK2V617F locus through mitotic amplification resulting in homozygosity of JAK2V617F somatic mutation on chromosome 9p (Table 2) [25]. Godfrey et al. studied the JAK2 mutation status of BFU-E grown in low erythropoietin conditions in 77 patients with PV or ET [45]. Using microsatellite PCR to map loss-of-heterozygosity breakpoints within individual colonies, homozygous JAK2V617F mutant colonies were absent or present in low percentages in heterozygous ET, but prevalent and common in patients with JAK2V617F-positive PV45. PV was distinguished from ET by expansion of a dominant homozygous JAK2V617F subclone, the selective advantage of which is likely to reflect additional genetic or epigenetic lesions. Hetero/homozygous or homozygous JAK2V617F mutation is associated with pronounced constitutively activation and genetic instability of megakaryopoiesis, erythropiesis and granulopoiesis in the bone marrow as the cause of hypercellular trilinear PV with a high risk of myelofibrosis (Table 2).
| Clinical and molecular criteria | Bone marrow pathology criteria (WHO) |
| ET | Normocellular ET |
| 1.Platelet count of >350 x109/l and the presence of large platelets in a blood smear 2.Heterozygous JAK2V617Fmutation, low mutation load 3.Normal erythrocytes <5.8x1012/L males, <5.6 x1012/L females, Hemoglobin(Hb) and hematocrit (ht) normal or upper range of normal |
Proliferation and clustering of enlargedmature pleomorphic megakaryocytes with hyperlobulated nuclei and maturecytoplasm, lacking conspicuous morphological abnormalities. Normocellular bone marrow (<60%) and no proliferation or immaturity of granulopoiesis orerythropoiesis. Reticuline fibrosis (RF) 0 or 1 |
| Prodromal PV | ET with bone marrow features of PV |
| 1. Platelet count of >350 x109/l Hb and Ht normal or in the upper range of normal, normal erythrocyte <5.8x1012/L males, <5.6x1012/L females. 2.Presence of JAK2V617F mutation 3.Low serum EPO level, increased LAP scoreand spontaneous EEC. |
Increased cellularity (60-80%) due to increased erytropoiesis or trilineagemyeloproliferation (i.e. panmyelosis). Proliferation and clustering of medium sized to lsarge (pleomorphic) msature megakaryocytes. Absence bone marrow features consistent with congenital polycythemia and secondary erythrocytosis. RF 0 or 1 |
| Prefibrotic hypercellular ET | EMGM |
| 1.Platelet count of>350 x109/l, 2.Presence of JAK2V617Fmutation and high JAK2 mutation load 3.Slight or moderate splenomegalyon ultrasound or on palpation 4.No preceding or alliedCML, PV, PMGM, RARS-T or MDS . Clinical stage 1: No anemia with Hb and Ht in the normal or low normal range: hb>12 g/dl, normal LDH and CD34+ Clinical stage 2: slight anemiaHb<12 to >10 g/dL, LDH↑, and splenomegaly Clinical stage 3: anemia, Hb<10 g/dL, LDH↑↑, CD34+ , leukoerythroblastose and, tear drop erythrocytes |
Hypercellular ET due to chronic megakaryocytic and granulocytic myeloproliferation (EMGM) and normal or reduced erythroid precursors. Loose to dense clustering of more pleiomorphic megakaryocytes with hyperploid or clumpsy nuclei (not or some cloud-like). Grading of reticulin fibrosis and MF in EMGM Prefibrotic: RF- 0/1=MF-0, no/minor splenomegaly Early fibrotic EMGM: RF 2=MF 1 and minor or moderate splenomegaly Fibrotic EMGM: RF3, RCF=MF2 andovert splenomegaly Post-ET MF: RF3/4=MF-2/3 (WHO criteria) |
Table 3: 2014 WHO Clinical Molecular and Pathobiological (WHO-CMP) criteria for diagnosis of JAK2V617F mutated essential thrombocythemia (ET) [32,33].
| Clinical and molecular criteria | Bone marrow pathology criteria (WHO) |
| Major criteria for PV A 1. Hematocrit>0.51/>0.48 in male/female Erythrocytes >5.8x1012/Lmales >5.6x1012/L females A 2. Presence of heterozygous and/or homozygous JAK2V617F or JAK2 exon 12 mutation A 3. Low serum Epo level Minor B 1. Persistent increase of platelet count x109/L: grade I: 400-1500, grade II: >1500. B 2. Granulocytes >10 x109/l or Leukocytes >12 x109/l and raised LAP-score or increased CD11b expression in the absence of fever or infection B 3. Splenomegaly on ultrasound echogram (>12 cm length in diameter) or on palpation. B 4. Spontaneous endogenous erythroid colony (EEC) formation (optional) |
P1. Bone marrow pathology: increased cellularity (60-100%) due to trilinearincrease of erythropoiesis, megakaryopoiesis and granulopoiesis and clustering of small to giant (pleomorph) megakaryocytes with hyperlobulated nuclei. Absence of stainable iron. No pronounced inflammatory reaction P2. Erythrocytosis. Normal erythropoiesis, normal granulopoiesis and megakaryocytes of normal size, morphology and no clustering Grading of reticulin fibrosis (RF) and reticulin collagen myelofibrosis (MF) Prefibrotic:RF-0/1=MF-0 Early fibrotic: RF-2=MF-1 Fibrotic:RCF 3=MF-2 Post-PV MF:RF 4=MF-3 |
Table 4: 2014 WHO Clinical Molecular andPathological (WHO-CMP) criteria for the diagnosis of prodromal, masked and classical JAK2 mutated polycythemia vera (PV) versus primary or secondary erythrocytoses [32,33].
| Clinical and molecularJAK2 wild type ET | Bone marrow pathology criteria (WHO) |
| 1.Platelet count >350x109/L and presence of large platelets in blood smear 2.Hemoglobin, haematocrit and erythrocyte count in the normal range 3.Presence of MPL515mutation and JAK2 wild type 4.Normal serum EPO 5.Normal LAP score and CD11b expression 6.No or slight splenomegaly 7.No leukoerythroblastosis 8.No preceding or allied CML, PV, RAS-T or MDS |
P1 . Proliferation of large to giant mature megakaryocyte with hyperlobulated, staghorn-like nuclei in a normocellular bone marrow (<65%) No increase of erythropoiesis, and no increase or immaturity of granulopoiesis or erythropoiesis, No or slight increase in reticulinRF 0/1 ET à MF Increased reticulin fibrosis around dense clustered megakaryocytes in a normocellular bone marrowand reduced erythropoiesis. Follow-up data of RF and MF related to splenomegaly in MPL 515 ET transltional states to MF are lacking. Grading of reticulin fibrosis (RF) and myelofibrosis (MF)similar as described for PV |
Table 5: 2014 WHO Clinical Molecular andPathological (WHO-CMP) criteria for the diagnosis of normocelular ET carrying one of the MPL515 mutations [32].
According to WHO [30] and WHO-CMP criteria (Tables 3 and 4) [32,33], heterozygous JAK2V617F positive ET is defined by a normocellular bone marrow histology with slight increase of erythropoiesis (Figure 2) or with a hypercellular bone marrow histology due to increased erythropoiesis (prodromal PV or masked PV). JAK2V617F positive hypercellular ET associated with prefibrotic megakaryocytic granuloctic myeloproliferation and (relative) reduction of erythropoiesis is consistent with hypercellular ET associated with a MGM bone marrow (EMGM or masked PV, Table 3) [32,33]. JAK2V617F mutated WHO defined PV typically shows a hypercellular bone marrow histology due to increased trilinear hematopoiesis of megakaryopoiesis, erythropoiesis and granulopoiesis (panmyelosis of Dameshek [34]) and no or slight increase of reticuline fibers (Figure 3 and Table 4).
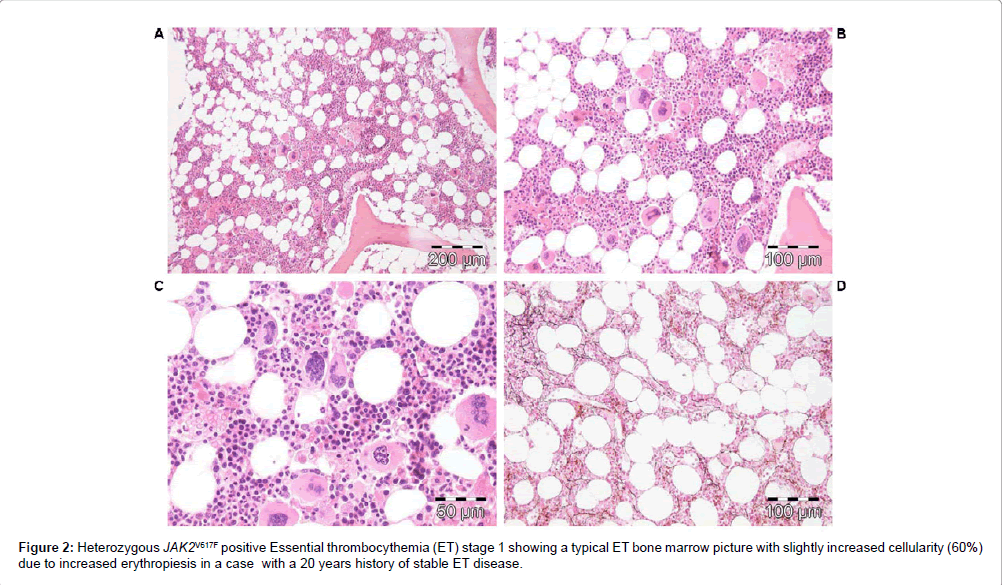
Figure 2: Heterozygous JAK2V617F positive Essential thrombocythemia (ET) stage 1 showing a typical ET bone marrow picture with slightly increased cellularity (60%) due to increased erythropiesis in a case with a 20 years history of stable ET disease.
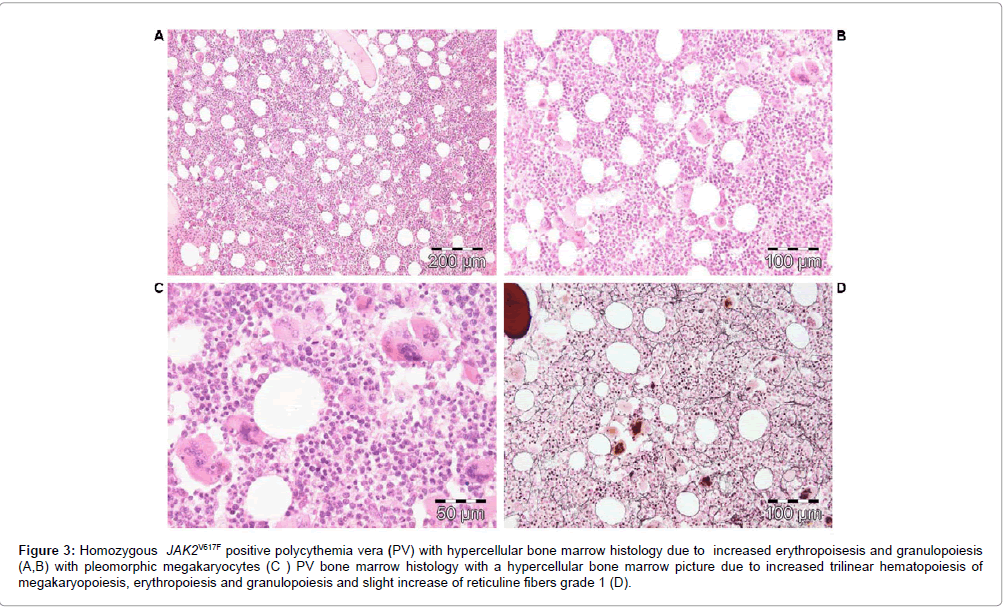
Figure 3: Homozygous JAK2V617F positive polycythemia vera (PV) with hypercellular bone marrow histology due to increased erythropoisesis and granulopoiesis (A,B) with pleomorphic megakaryocytes (C ) PV bone marrow histology with a hypercellular bone marrow picture due to increased trilinear hematopoiesis of megakaryopoiesis, erythropoiesis and granulopoiesis and slight increase of reticuline fibers grade 1 (D).
The UK MPN Study Group assessed the clinical features in the cohort of 806 PVSG defined ET patients subdivided in 414 JAK2V617F positive and 362 JAK2 wild type ET and evaluated the bone marrow features in 393 ET patients [46,47]. JAK2V617F positive ET patients had multiple features of PV such as a significantly higher hemoglobin, lower serum EPO and ferritin, higher neutrophils, bone marrow erythrocytosis and granulocytosis, more venous thrombosis and a higher rate of polycythemic transformation. PVSG defined JAK2 wild type ET had significant higher platelet counts (962, range 668-1535x109/L) than JAK2V617F-positive ET (846, range 632-1222x109/L) [46]. In the UK MPN (Primary Thrombocythemia 1 (PT-1) study, bone marrow trephine of 209 JAK2V617F positive and 184 JAK2 wild type ET was independently assessed by 3 hematopathologists who did not know the JAK2 mutation status [47]. The overall cellularity was significantly increased in JAK2V617F mutated ET as compared to JAK2 wild type ET, indicating that increased erythroid and/or granulocytic cellularity are features of prodromal PV or masked PV [47].
Pich et al. prospectively analyzed histological changes in diagnostic bone marrow biopsy from 2006-2010 of 103 newly diagnosed WHO defined ET patients [48]. Bone marrow features in 44 JAK2 wild ET cases revealed prominent clusters of large megakaryocytes with staghorn nuclei, less micromegakaryocytes and no or minor erythroid hyperplasia as compared to JAK2V617F positive ET [48]. In contrast, 59 JAK2V617F positive ET patients revealed a typical PV bone marrow histology with higher hemoglobin, hematocrit, erythrocytes and increased bone marrow due to hyperplasia of erythroid and myeloid lineages and the presence of pleomorphic megakaryocytes very similar as in WHO-CMP defined ET and PV (Tables 3 and 4). The mean and median JAK2V617F mutation burden in 2008 WHO defined ET was 14.4% and 8.7% respectively [48]. Interestingly LDH (604+132) and spleen size (15.4+4.9 cm on echogram) in 16 ET cases with a JAK2V617F mutation load above 12.5% were significantly increased as compared to normal LDH (386+94) and normal spleen size (11.2 ± 2.1 cm on echogram) in 37 ET cases with a JAK2V617F mutation load below 12.5% [48].
JAK2 exon 12 mutations as cause of IE and PV
The finding of the JAK2 exon 12 mutations in PV patients negative for the JAK2V617F mutation further confirms the strong association between the JAK2 mutations and MPN [49,50]. The 5% PV patients negative for JAK2V617F are frequently heterozygous for exon 12 JAK2 mutations and usually present with early stage PV or idiopathic erythrocythemia (IE) with favourable outcome and normal life expectancy. The UK MPN Study Group identified JAK2 exon 12 mutations in 10 JAK2V617F – negative PV patients with increased red cell mass, which according to PVSG criteria could diagnosed as PV in 6 and IE in 4 cases [49]. Pre-treatment bone marrow biopsies in 5 patients carrying one of the JAK2 exon 12 mutations showed characteristic erythroid hyperplasia with some morphological abnormalities of the megakaryocyte and normal granulopoiesis in bone marrow biopsy specimens clearly different from primary or secondary erythrocytosis. In the Mayo Clinic study [50], 5 cases of JAK2V617F negative PV carrying JAK2 exon 12 mutation (F537-K539delins or N542-E534del) were diagnosed as IE with increased hemoglobin and hematocrit, low serum EPO, normal platelet and leukocyte counts, no or palpable spleen and a typical hypercellular bone histopathology predominantly due to erythroid hyperplasia and clusters enlarged megakaryocytes with hyperploid nuclei was observed in 2 cases [51]. The bone marrow histology by Lakey et al. in 7 JAK2 exon 12 mutaed MPN patients (IE in 4, PV in 2, MF in 1) revealed hyperplasia of atypical small to medium-sized large megakaryocytes was present in all (Figure 4) [51], which differs from JAK2V617F mutated PV (Figure 3). The JAK2 exon 12 MPN cases lack the prominent clusters of large megakaryocytes with hyperlobulated nuclei that characterize JAK2V617F-positive prodromal and classical PV (Figure 3). A spectrum of small to medium sized megakaryocyte is seen in JAK2 exon 12 PV bone marrows with a predominance of smaller forms with atypical nuclei with various degrees of monolobation to hyperlobation and abnormal chromatin distribution (Figure 4) [51]. Bone marrow reticulin fibrosis was normal or slightly increased in 6, and one case evolved 15 years after intitial diagnosis into post-PV myelofibrosis with reticulin fibrosis grade 3 [51]. The JAK2 exon 12 IE or PV patients presented aquagenic pruritis and/or erythromelalgia in 3 and microvascularevents including headache, dizziness, blurred vision and distal extremity numbness (aspirin responsive platelet thrombophilia or Sticky Platelet Syndrome) [4,5] in 4 at platelet counts between 152 and 790 x 109/L (of whom 5 below and 2 above 300 x 109/L). The hemoglobin ranged from 18.3 to 22 g/dL and leukocytes were below 10.5 x 109/L in 6 of 7 JAK2 exon 12 PV cases consistent with the diagnosis of idiopathic erythrocytosis (IE) [51]. Serum EPO levels were low and karyotyping was normal in all 7 JAK2 exon12 MPN cases.
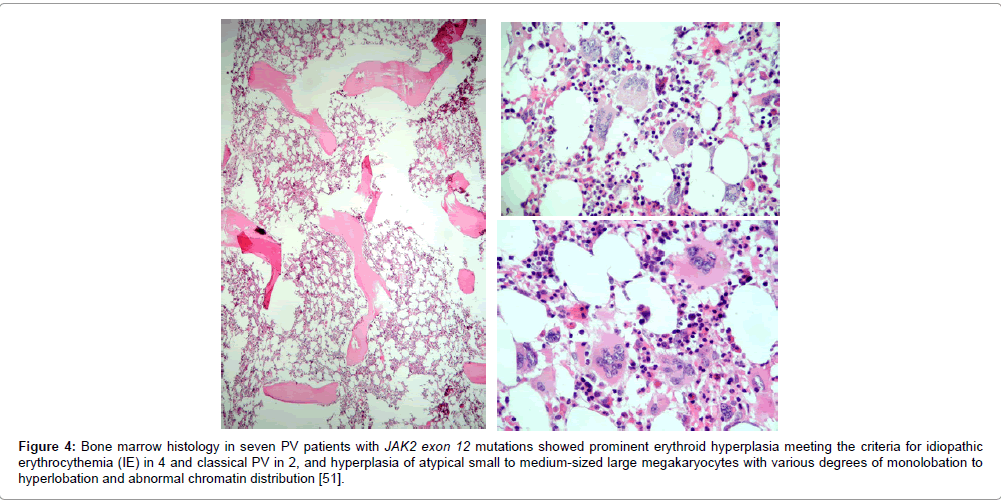
Figure 4: Bone marrow histology in seven PV patients with JAK2 exon 12 mutations showed prominent erythroid hyperplasia meeting the criteria for idiopathic erythrocythemia (IE) in 4 and classical PV in 2, and hyperplasia of atypical small to medium-sized large megakaryocytes with various degrees of monolobation to hyperlobation and abnormal chromatin distribution [51].
JAK2 wild type ET and MF carrying the MPL515 mutation
Congenital ET due to a gain of function mutation in the cMPL gene has been described in 2004 [52]. This led to the search and discovery of the MPLW515L and MPLW515K mutations as the driver cause of ET and myelofibrosis (MF) [53-55]. Within the JAK2 wild type MPN, there is a small subgroup who carry an acquired gain of function mutation of the MPL receptor as the cause of ET: 3% in the Italian study [55], and 8.5% in the UK studies [56,57]. In contrast to JAK2V617F mutated trilinear MPN, MPL515 mutated ET patients have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis, do not evolve into overt PV during follow-up, have normal serum EPO and ferritin levels, and show pronounced megakaryocytic proliferation of small and large (giant) mature megakaryocytes and no increase of erythropoiesis in the bone marrow (Table 5) [33]. In 2014 we produced good evidence that JAK2 wild type ET carrying the MPL515 mutation typically present with normacellular ET histology with the presence of giant megakaryocyte with hyperobulated staghoen-like nuclei and no increase of erythropoiesis clearly differ from JAK2V617F mutated ET (Table 5) [33]. As compared to bone marrow histopathology in JAK2V617F mutated ET (Figure 2) there were significant differences on three points. First, the megakaryocytes in MPL515 mutated PT are larger than in PV (Figure 5). In contrast, the megakaryocytes in JAK2V617F mutated ET are not larger than in PV and show similar pleomorphic megakaryocytes morphology as in PV (compare Figures 2 and 3). Second, there was local increase of erythropoiesis in areas of loose clustered pleiomorphic megakaryoctyes in JAK2V617F mutated ET (Figure 2), but not in MPL515 mutated ET (Figure 5). Third, we observed increased reticulin fibers grade 2 in a rather normocellular bone marrow in areas of dense clustered megakaryocytes in MPL515 mutated ET, which is not seen in JAK2V617F mutated normocellular ET, hypercellular prodromal PV and early stage PV [33]. Whether such differences of megakaryocyte morphology in bone marrow biopsies are characteristic enough to distinguish normocellular JAK2V617F mutated ET with low JAK2 mutation load (Table 3) from MPL515 mutated ET and MF (Table 5) respectively by expert hematopathologists remains to be evaluated in large prospective clinical and basic research studies of newly diagnosed and previously untreated MPN patients.
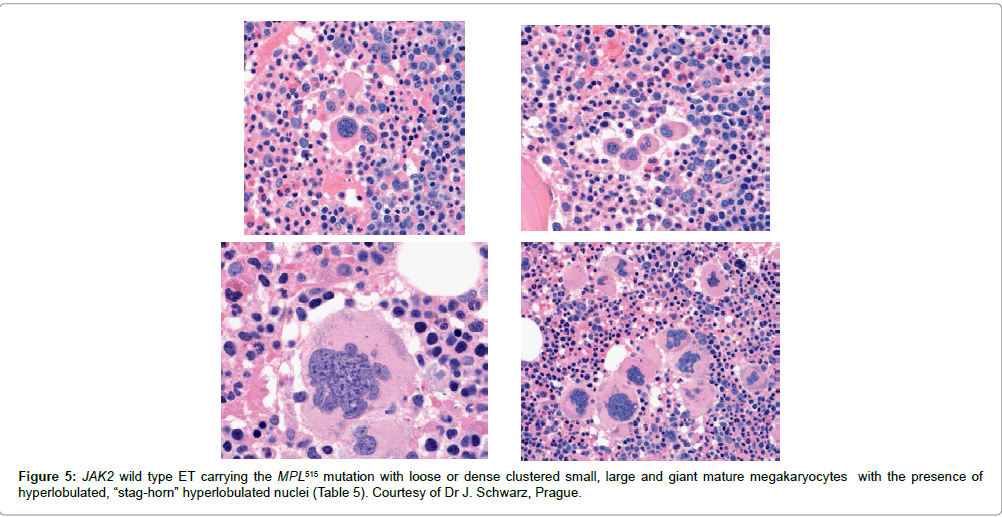
Figure 5: JAK2 wild type ET carrying the MPL515 mutation with loose or dense clustered small, large and giant mature megakaryocytes with the presence of hyperlobulated, “stag-horn” hyperlobulated nuclei (Table 5). Courtesy of Dr J. Schwarz, Prague.
Calreticulin (CALR) mutated ET and MF
The molecular etiology of JAK2/MPL wild type ET and MF remained elusive untill Kralovics in Vienna and Green in the UK independently discovered in 2013 the calreticulin (CALR) mutations in JAK2 wild type ET and MF patients [8,58]. Dr Kralovics and his team in Austria and Italy described the occurrence of CALR mutation in 78 of 311 (25%) ET patients and in 72 of 203 (35%) MF patients and in none of 382 PV patients [58]. CALR mutations are mutually exclusive with both JAK2V617F and MPL515 mutations. All ET and MF patients with CALR in exon 9 are JAK2 and MPL wild type. Out of a cohort of 289 JAK2 wild type ET 195 (67%) carried one of the CALR mutations. Out of a cohort of 120 JAK2 wild type MF a CALR mutation was detected in 105 (80%). In the 150 patients with the CALR mutation for whom matched T-lymphocyte DNA was available, the CALR mutations were somatic. In none of 45 CML, 73 MDS 64 CMML and 24 RARS-T (refractory anemia with increased ringed sideroblast and thrombocytosis) patients the CALR mutation was not found except that 3 SF3B1 positive RARS-T patients with myelodysplasia carried a CALR mutation. A total of 36 types of somatic mutations (insertions and deletions) were detected in exon 9 of the CALR gene encoding the C-terminal amino acids of CALR protein and only 3 patients were homozygous. In the total cohort of 1235 ET and MF patients 63.4%, 4.4% and 23.5% carried the JAK2V617F, MPL515 and CALR mutation respectively, and 8.8% were triple negative for these clonal driver mutation [58]. Green and his team in the UK independently found somatic CALR mutations in 70 to 84% of MPN samples with nonmutated JAK2 ET or MF [59]. CALR exon 9 mutations were found in 26 of 31 (84%) patients with ET or MF and nonmutated JAK2. CALR exon 9 mutations were absent in all 120 patients who had JAK2 or MPL mutations. CALR mutations were present in 110 of 158 MPN patients lacking JAK2 or MPL, including 80 of 112 (70%) ET patients, 18 of 32 (56%) MF patients and 12 of 13 patients with progression of ET to MF. CALR mutations were identified in 10 of 120 (8%) MDS patients (RA in 5 of 53, RARS in 3 of 27 and RAEB-T in 2 of 27), and in one patient each with chronic myelomonocytic leukemia (CMML) and atypical CML. No CALR mutations were found in control samples, lymphoid cancers, solid tumors, or cell lines [59]. Overall, CALR exon 9 mutations were identified in 148 patients. All CALR mutations were indels with 19 distinct variant: 14 deletions, 2 insertions and 3 complex indels, which generated a +1 base-pair frameshift, which would result in a mutant with a novel C-terminal [59].
According to our experiences bone marrow histology in prefibrotic and early fibrotic MPN in JAK2 wild type hypercellular ET of subtype PMGM show dysmorphic megakaryocytes with definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio (Table 6), which are not seen in JAK2 wild type ET carrying the MPL515 mutation and also not in prefibrotic JAK2V617F mutated ET, ET/PV and PV [32,33]. In six consecutive previously untreated CALR mutated ET and MF patients we found that the bone marrow histology findings in CALR mutated ET and early MF were consistent with hypercellular ET as the presenting feature of prefibrotic and early fibrotic stages of PMGM (Figures 6 and 7). The bone marrow histology findings in these 6 consecutive CALR mutated ET and MF patients showed a typical PMGM picture (Table 6), which were significantly different from giant megakaryocytes and hyperlobulated staghorn-like nuclei in MPL515 mutated ET (Figure 5), in JAK2 exon 12 mutated IE or PV (Figure 4) and in JAK2V617F mutated ET and PV (Figures 2 and 3). The WHO-CMP featurres of a large group of CALR mutated ET and MF patients (N=50) as compared to a group of 50 cases with JAK2V616F positive ET, prodromal PV and PV at time of first diagnosis are currently under investigation.
| Clinical criteria JAK2 wild type PMGM | Pathological criteria of CALR MGM |
| A1 No preceding or allied other subtype of myeloproliferative neoplasm PV, CML, MDS. The main presenting features is pronounced isolated thrombocythemia with platelet count around or above 1000x109/L A2 Presence of CALR mutation and JAK2 wild type C Clinical stages of CALRMGM C 1. Early clinical stage: Hb>12g/dL, slight to moderate splenomegaly, thrombocytosis around or above 1000x109/L, normal LAP score C2. Intermediate clinical stage: slight anemiaHb<12 to >10 g/dL, decreasing platelet count, splenomegaly, increased LDH and definitive tear drop erythrocytes C3. Advanced stage: anemiaHb<10 g/dL, tear drop erythrocytes, increased LDH, increased CD34+ cells, pronounced splenomegaly, normal or decreased platelet counts, leucocytosis or leukopenia. |
P1 Primary megakaryocytic granulocytic myeloproliferation (PMGM) and relative or absolute reduction of erythropoiesis and erythroid precursors. Abnormal dense clustering and increase in atypical medium sized, large to giant immature megakaryocytes containing bulbous (cloud-like) hypolobulated nuclei and definitive maturation defects MF Grading reticulin fibrosis (RF), and reticulncolloagenmyelofibrosis (MF) MF 0 PrefibroticCALR MGM, no reticulin fibrosis RF 0/1 MF 1 Early fibrotic CALR MGM slight reticulin fibrosis RF 2 MF 2 Fibrotic CALR MGM increase RF grade 3 and slight to moderate collagen fibrosis MF 3 Advanced fibrotic CALR MGM with collagen fibrosis-osteosclerosis |
Table 6: 2014 WHO-CMP criteria for hypercellular ET associated with primary megakaryocytic, granulocytic myeloproliferation (PMGM) [25-28,32] carrying on of the
calreticulin (CALR) mutations [33].
With the advent of the JAK2V617F mutation latent, masked, early and overt stages of PV will be picked up more than 5 to 10 years earlier by the WHO-CMP criteria as compared to the PVSG and WHO criteria. A broad spectrum of heterozygous JAK2V617F mutated ET, hetero/ homozygous JAK2V617F mutated masked PV, classical PV and post- ET MF or post-PV MF is very characteristic for JAK2V617F mutated trilinear MPN as the main distinct and most frequent MPN disease entity (Tables 3 and 4). JAK2 wild type ET and MF carrying one of the MPL515 mutations is the second distinct MPN without features of PV at diagnosis and during follow-up (Table 5). The CALR mutated ET and MF category became in 2013/2014 the third distinct MPN entity without features of PV (Table 6). CALR mutations were found in a few MDS (RARS-T) patients, very rarely in atypical CML or CMML patients, but not in JAK2V617F mutated ET and PV patients and also not in BCR/ABL positive CML patients [58,59]. CALR patients had no features of PV with lower hemoglobin and white blood cells counts but higher platelet counts [60,61]. The lower incidence of thrombotic complications in CALR mutated thrombocythemia has been attributed to the fact that CALR-positive ET and MF patients lack PV features with significantly lower hemoglobin and WBC values than those in JAK2V617F mutated ET and prodromal PV patients [60,61]. Patients with JAK2V617F mutated ET and PV had a similar high risk of major thrombosis, which was twice that of CALR utated thrombocythemia patients [60,61]. CALR mutated ET patients presented with higher platelet counts and lower hemoglobin levels similar as observed in PMGM [31] as compared to hemoglobin levels in the upper range of normal in JAK2 mutated MPN patients (prodromal PV) [60,61]. WHO-CMP features of PV and polycythemic transformation has never been observed both in PMGM [26,31] and in CALR mutated ET and MF patients [60,61]. The evolution of ET to MF belong to the natural history of all molecular variants of the MPNs, which appeared to be related to the acquisition of epigenetics events on top of the driver mutation JAK2, MPL or CALR [62,63]. Life expectance was significantly longer in CALR mutated MF patients as compared to those with a JAK2V617F, which can be explained by the fact that CALR mutated MPN patients were about 10 years younger at time of diagnosis [60-62]. The overall survival of CALR mutated MF patients in the Italian studies was 23 years as compared to 14.4 years of MF patients with the JAK2V617F mutated MF.
Retrospective analysis of 254 WHO-defined MF patients in the Mayo Clinics, Rochester showed that the JAK2-, MPL- and CALRmutations were detected in 58%, 8.3 and 25% respectively and 8.7% were triple negative [62]. The median overall survival (OS) among 253 WHO-defined PMF patients in 83 CALR-, 21 MPL-, and 147 JAK2- mutated cases and in 22 triple negative cases was 8.2, 4.1, 4.3 and 2.5 years. As compared to CALR wild type MF, CALR-mutated MF patients were younger, had higher platelet count, lower leukocyte count, higher hemoglobin (less anemic) and lower DIPSS-plus score. CALR-mutated MF patients had a favorable impact on median survival as compared to CALR-negative MF patients whether ASXL1-negative or positive. Among 181 WHO-defined CALR-negative MF patients, the median overall survival was 2.3 years in 55 CALR-negative/ASLX1-positive as compared to 5.6 years in 126 CALR-negative/ASXL1-negative MF patients. Among 146 CALR-positive MF cases the median survival was 7 years in 20 CALR-positive/ASXL1-positive MF patients as compared to 9.6 years in 126 CALR-positive/ASXL1-negative MF patients [62]. The etiology of triple JAK/MPL/CALR negative MF remains elusive wheher they represent MPN or MDS. The awareness of the molecular heterogeneity of the MPNs including JAK, MPL and CALR mutations on top of epigenetic factors reflect the funeral of the term primary myelofibrosis (PMF)63. The terms CIMF and PMF according to the 2001and 2008 WHO classifications should be replaced by 2014 WHOCMP defined CALR-, MPL515-mutated ET and MF (Tables 5 and 6) and JAK2V617F-mutated ET, PV and MF (Tables 3 and 4) with variable degrees of anemia, splenomegaly, hypersplenism and myelofibrotic transformation of the bone marrow. Grading of reticulin fibrosis (RF) and reticulin collagen myelofibosis (MF) is a second event in all variant of JAK2, MPL and CALR mutated MPNs (Tables 3-6 and Figures 8-10). In the congenital hereditary ET (HET) caused by gain of function mutation in the TPO and JAK2 gene ( JAK2V617I and JAK2 R564Q) the responses of mutated CD33 and CD34+ cells to TPO are increased, but the responses to EPO were normal thereby explaining why HET caused by heterozygous germline TPO and JAK2 mutations are associated with the biological and clinical characteristics of ET without PV features (Figure 10) [64-66]. The clinical manifestations of autosomal dominant HET are typically complicated by Platelet Sticky Syndrome (ASPS), a novel disease entity of congenital aspirin responsive platelet thrombophilia [64-66].
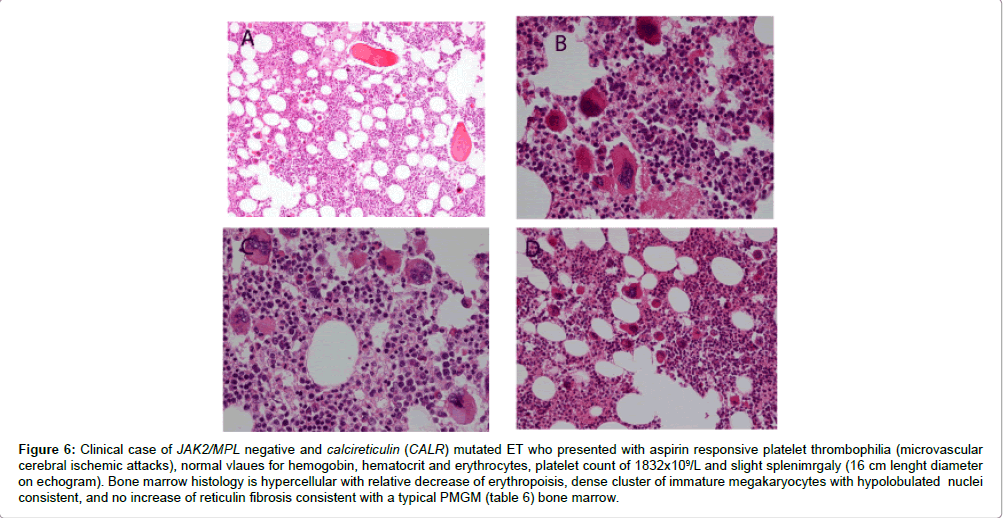
Figure 6: Clinical case of JAK2/MPL negative and calcireticulin (CALR) mutated ET who presented with aspirin responsive platelet thrombophilia (microvascular cerebral ischemic attacks), normal vlaues for hemogobin, hematocrit and erythrocytes, platelet count of 1832x109/L and slight splenimrgaly (16 cm lenght diameter on echogram). Bone marrow histology is hypercellular with relative decrease of erythropoisis, dense cluster of immature megakaryocytes with hypolobulated nuclei consistent, and no increase of reticulin fibrosis consistent with a typical PMGM (table 6) bone marrow.
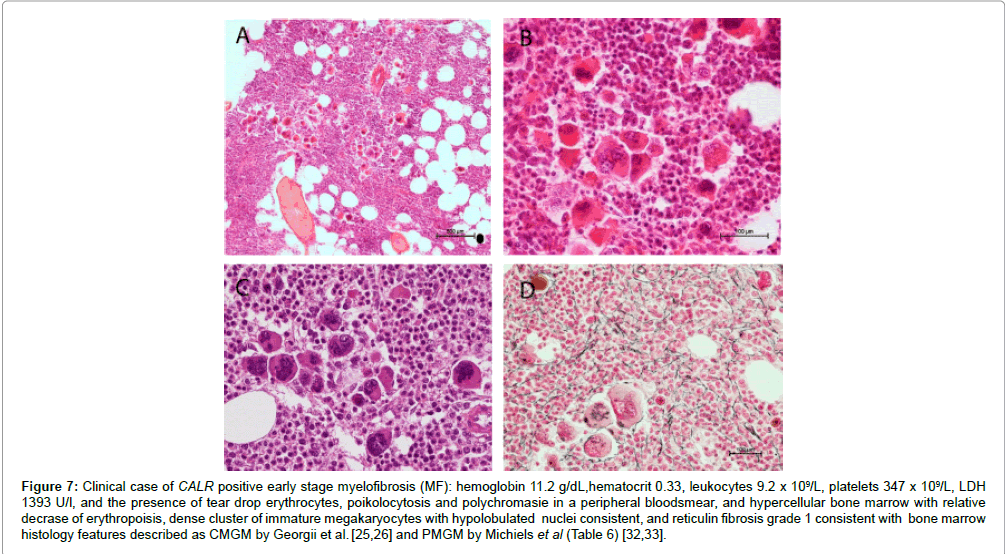
Figure 7: Clinical case of CALR positive early stage myelofibrosis (MF): hemoglobin 11.2 g/dL,hematocrit 0.33, leukocytes 9.2 x 109/L, platelets 347 x 109/L, LDH 1393 U/l, and the presence of tear drop erythrocytes, poikolocytosis and polychromasie in a peripheral bloodsmear, and hypercellular bone marrow with relative decrase of erythropoisis, dense cluster of immature megakaryocytes with hypolobulated nuclei consistent, and reticulin fibrosis grade 1 consistent with bone marrow histology features described as CMGM by Georgii et al. [25,26] and PMGM by Michiels et al (Table 6) [32,33].
We conclude that Bone Marrow Pathology (BMP) is a pathognomonic clue to each of the JAK2, MPL and CALR mutated MPN and clearly distiguishes all variants of MPN from CML, RARS-T, and MDS including the 5q inus syndrome mainly based on megakaryocyte morphology with a sensitivity and specificity of near to 100% (Figures 8-10) [67,68]. Within the JAK2 mutated ET and PV at time of diagnosis the megakaryocytes are similar large and pleiomorphic with normocellular bone marrow in heterozygopus JAK2 mutated ET and increased cellularity of 60-80% due to increased erythropoiesis=prodromal PV and strongly increased 90-100% cellularity in more advanced EMGM=masked PV and in classical prefibrotic PV due to increased trilinear hematopoiesis of increased erythropoiesis, megakaryooiesis and granulopoisesis (Tables 3 and 4) [14,27-35,67,68]. Staging of MPN disease burden in the trilinear MPN phenotypes of JAK2 mutated ET, masked PV and overt PV is based on grading of splenomegaly, LDH, leukocyte count and grading of reticulkin fibrosis on top of JAK2 allele burden (Figures 8-10). Exon 12 JAK2 mutated PV reflects a more favorable presentation of mainly erythrocythemia and and early stage PV with no or slight increase of platelets and leukocytes during long-term follow-up as compared to JAK2V671F mutated masked and classical PV. JAK2 wild type ET carrying the MPL or CALR mutation do not show PV features in blood and bone marrow during the prefibrotic and early stages of reticulin fibrosis (myelofibrosis=MF) at diagnosis and during longterm follow-up. CALR mutated ET present with high platelet counts around 1000x109/L which after longterm follow-up tend to decline, which is related to progressive splenomegaly and increased MF (Figures 8-10). CALR mutated ET and MF is featured by hyeprcellular bone marrow due to dual primary megakaryocytiv granulocytic myeloproliferation (PMGM) with relative or absolute reduction of erythropoiesis and the presence of loose to dense clustered more or less immature large megakaryocytes with immature cloud-like nuclei, which are not seen in JAK2 mutated ET and PV and also not in MPL mutated ET (Figures 8-10). Bone Marrow Pathology (BMP) has a specificity and sensitivity near to 100% to differentiate between all molecular variants of the the MPNs ET, PV and early stage MF from reactive thrombocytosis and primary or secondary erythrocytoses [67-70]. Bone marrow histology alone has a near to 100% accuracy to distinct MPN from CML and MDS (RARS-T and 5q-minus syndrome). The sensitivity and specificity to distinguish JAK2 on one hand vs MPL vs CALR mutated ET on the other hand just based on bone marrow histology alone surely will not always be possible [69,70]. The distinction of JAK2, versus MPL versus CALR mutated ET based on megakaryocyte morphology and backgound increase of cellularity is predicted to have an estimated accuracy of 70 to 80% in pretreatment diagnostic bone marrow biopsies at time of diagnosis [67-70]. The diagnostic differentiation and staging of the early and fibrotic stages of JAK2, MPL and CALR mutated MPNs should be based on bone marrow morphology on top of quantitaive measurement of JAK2, MPL and CALR mutation load related to the degree of anemia and splenomegaly to validate the natural history of each of the MPN disease burden prospectively during evolution of all molecular variants of ET into MF [25-28,67-70].
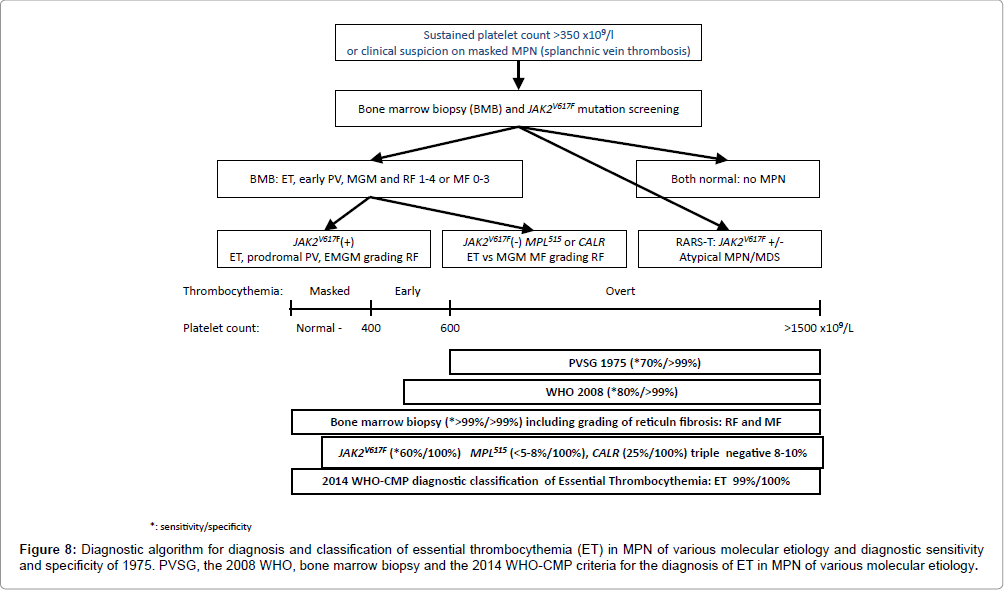
Figure 8: Diagnostic algorithm for diagnosis and classification of essential thrombocythemia (ET) in MPN of various molecular etiology and diagnostic sensitivity and specificity of 1975. PVSG, the 2008 WHO, bone marrow biopsy and the 2014 WHO-CMP criteria for the diagnosis of ET in MPN of various molecular etiology.
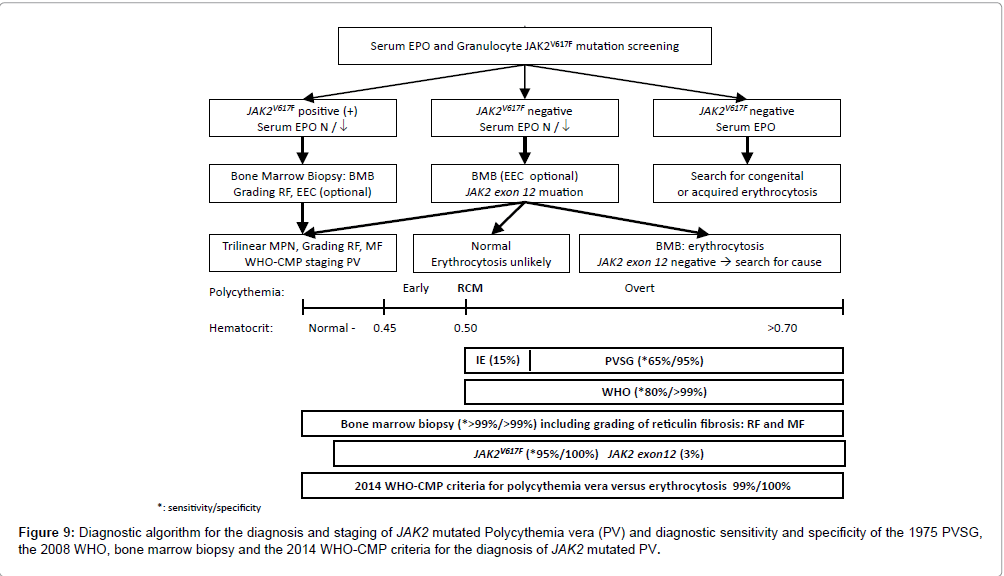
Figure 9: Diagnostic algorithm for the diagnosis and staging of JAK2 mutated Polycythemia vera (PV) and diagnostic sensitivity and specificity of the 1975 PVSG, the 2008 WHO, bone marrow biopsy and the 2014 WHO-CMP criteria for the diagnosis of JAK2 mutated PV.
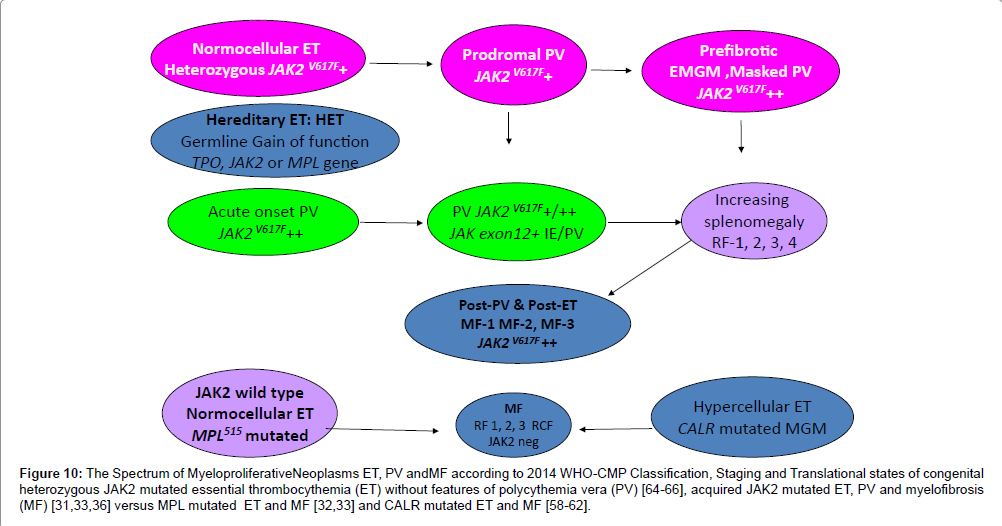
Figure 10: The Spectrum of MyeloproliferativeNeoplasms ET, PV andMF according to 2014 WHO-CMP Classification, Staging and Translational states of congenital heterozygous JAK2 mutated essential thrombocythemia (ET) without features of polycythemia vera (PV) [64-66], acquired JAK2 mutated ET, PV and myelofibrosis (MF) [31,33,36] versus MPL mutated ET and MF [32,33] and CALR mutated ET and MF [58-62].