Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2020)
The COVID-19 pandemic has caused greater deaths worldwide and is continuing to do so. India is no exception to this and the number of positive cases in India at increasing at an alarming rate. With no vaccine or medicine to either prevent or cure it, the medical fraternity and people worldwide are relying on basic precautionary measures. There is an utmost need of a drug to cure the pandemic. Various studies have been reported for the genomic and proteomic structure of the virus SARS-CoV-2, which causes the COVID-19. There are many important proteins in the viral genome which can be targeted for drug design and discovery. The current paper focuses on Main protease and RNA dependent RNA polymerase (RdRp) proteins for the drug design against COVID-19. Two important molecules such as Curcumin and Allicin having medicinal properties were studied for their antiviral activity against the SARS-CoV-2 proteins. Hexahydrocurcumin and Hexahydrourcuminol amongst the Curcumin derivatives were found to show good binding affinity against the Main protease; Dihydrocurcumin glucuronide and Curcumin sulphate showed good binding against RdRp. Amongst the Allicin derivatives, Allicin and Ajoene showed good binding affinity for both Main protease as well as RdRp protein. Thus these molecules can be further studied for their in vitro and in vivo activity testing against COVID-19.
SARS-CoV-2; Main protease; RNA dependent RNA polymerase; Allicin; Curcumin; In silico drug design; Docking
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the strain of coronavirus that causes coronavirus ailment 2019 (COVID-19), a respiratory sickness. It is infectious in people, and the World Health Organization (WHO) has assigned the progressing pandemic of COVID- 19 a General Wellbeing Crisis of Universal Concern. The COVID-19 pandemic in India is a piece of the overall pandemic of coronavirus sickness 2019 (COVID-19). The first instance of COVID-19 in India, which may have originated from China, was accounted for on 30 January 2020. As of 9 July 2020, the Ministry of Health and Family Welfare (MoHFW) has confirmed a total of 767,296 cases, 476,377 recoveries (including 1 migration) and 21,129 passings in the nation. Figure 1 shows the map of India with the active cases [1-4].

Figure 1: No of confirmed cases in India as on 9th July 2020
Every SARS-CoV-2 virion is 50–200 nanometers in diameter. Like different coronaviruses, SARS-CoV-2 has four auxiliary proteins, known as the S (spike), E (envelope), M (membrane), and N (nucleocapsid) proteins; the N protein holds the RNA genome, and the S, E, and M proteins together make the viral envelope. The spike protein, which has been imaged at the nuclear level utilizing cryogenic electron microscopy, is the protein answerable for permitting the infection to connect to and combine with the layer of a host cell; explicitly, its S1 subunit catalyzes connection, the S2 subunit combination [5].
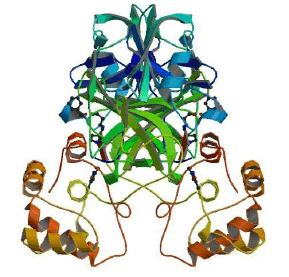
The SARS-CoV-2 Main protease and RNA dependent RNA polymerase are 2 important proteins of the virus involved in the replication and overall survival of the virus in the host cell. Thus these proteins are the main targets for current research and development against COVID-19. These are probable targets for design and development of drugs against the infection. The proteases play essential roles in cutting the polyproteins into all of the functional pieces. The main protease of coronavirus makes the majority of these cuts. It is a dimer of two indistinguishable subunits that together structure two dynamic destinations. The protein overlay is like serine proteases like trypsin, yet a cysteine amino corrosive and a close by histidine play out the proteincutting response and an additional area balances out the dimer. The structure of SARS-CoV-2 main protease is highlighted in Figure 2 [6].

Figure 2: SARS-CoV-2 main protease dimer with inhibitor in blue colour PDB id 6LU7.
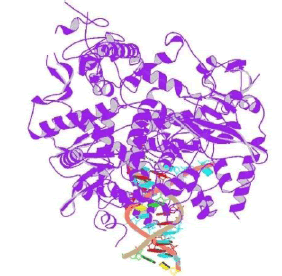
The RNA-dependent RNA polymerase (RdRp, also named nsp12), which catalyzes the synthesis of viral RNA, is a key component of coronaviral replication/transcription machinery and appears to be a primary target for the antiviral drugs such as remdesivir. The nsp12 RNA-dependent RNA polymerase possesses some minimal activity on its own6, but the addition of the nsp7 and nsp8 cofactors greatly stimulates polymerase activity. nsp12 contains a polymerase domain (a.a. 398–919) that assumes a structure resembling a cupped “right hand” similar to other polymerases. The polymerase domain is comprised of a fingers domain (amino acids (a.a.) 398– 581, 628–687), a palm domain (a.a. 582–627, 688–815), and a thumb domain. Figure 3 shows the structure of RNA dependent RNA polymerase [7,8].

Figure 3: SARS-CoV-2 RNA dependent RNA polymerase in complex with nsp7 and nsp8 PDB id 7BV2.
Thus these two important proteins were chosen as targets for the design and development of drugs against COVID-19. Two important naturally occurring compounds with medicinal properties were checked for their antiviral properties against SARS-CoV-2. Curcumin is a splendid yellow compound made by Curcuma longa plants. It is the major curcuminoid of turmeric (Curcuma longa), an individual from the ginger family, Zingiberaceae. It is sold as a natural supplement, beautifiers’ part, and in food seasoning, and food shading. Chemically, Curcumin is a diarylheptanoid, having a place in the family of curcuminoids, which are normal phenols liable for turmeric's yellow shading. It is a tautomeric compound existing in enolic structure in organic solvents and in keto structure in water. Elements that limit the bioactivity of Curcumin or its analogs incorporate chemical instability, water insolubility, absence of potent and selective target activity, low bioavailability, limited tissue distribution, and extensive metabolism. Next to no Curcumin gets away from the GI tract and most is discharged in excrement unaltered. In the event that Curcumin enters plasma in sensible sums, there is a high danger of harmfulness since it is indiscriminate, and communicates with a few proteins known to expand the danger of unfavorable impacts, including hERG, cytochrome P450s, and glutathione S-transferase.
Anyway During the most recent decade, analysts have thought of various different formulations with an attention on improving the bioavailability of Curcumin. Thus, a critical number of bioavailable Curcumin-based formulations were presented with the changing scope of upgraded bioavailability. For instance, piperine is the significant dynamic segment of dark pepper and, when consolidated in a complex with Curcumin, has been appeared to expand bioavailability by 2000%. Curcumin joined with improving specialists gives various medical advantages. Good tolerability and safety profiles have appeared in clinical trials, even at portions somewhere in the range of 4000 and 8000 mg/day and of dosages up to 12,000 mg/day of 95% centralization of three curcuminoids: Curcumin, bisdemethoxycurcumin, and demethoxycurcumin. Recent advances in micro- and nano-formulations of Curcumin with incredibly improved assimilation bringing about attractive blood levels of the dynamic types of Curcumin now make it conceivable to address a wide scope of potential applications, including pain management, and as tissue protective. Thus we chose Curcumin as our target drug molecule to check its activity against SARS-CoV-2. Figure 4 shows the 2D structure of Allicin and Curcumin used in the study.

Figure 4: (a) Structure of Curcumin, (b) Structure of Allicin.
Allicin is an organosulfur compound obtained from garlic, in the family Alliaceae. It was first isolated and studied by Chester J. Cavallito and John Feeds Bailey in 1944. At the point when new garlic is hacked or squashed, the chemical alliinase changes over alliin into Allicin, which is answerable for the fragrance of new garlic. The Allicin created is flimsy and rapidly changes into a progression of other sulfur-containing mixes, for example, diallyl disulfide. Allicin is a slick, somewhat yellow fluid that gives garlic its novel smell. It is a thioester of sulfonic corrosive and is otherwise called allyl thiosulfinate. Its organic action can be ascribed to the two -its cell reinforcement action and its response with thiolcontaining proteins. Allicin has been read for its capability to treat different sorts of various multiple drug resistance bacterial infections, as well as viral and fungal infections in vitro. Thus to understand the antiviral activity, Allicin and its derivatives were chosen as next drug molecule.
The below flowchart protocol depicts the procedure followed in the study (Figure 5).

Figure 5: Flowchart of Methodology.
Target protein preparation
The crystal structure of main protease with PBD id 6LU7 and of RdRp protein with PDB id 7BV2 were taken as the target proteins. In the autodock software they were prepared by removing water, adding hydrogen, adjusting the torsion angles, applying the default forcefields, computing Gasteiger charges, merging non-polar H; assigning Autodock 4 atom types. Stouten atomic solvation parameters are also designated to the receptors. The receptors were done converted to .pdbqt format.
For the Libdock and CDocker they were prepared using the Prepare protein module. The proteins were subjected to CHARMm forcefield, with protein dielectric constant set at 10, pH for protonation was 7.4, water and other residues were removed and hydrogens were added. The maximum loop length was fixed at 20 and ionic strength of 0.145. The energy minimized structures of the proteins were taken for further study.
Ligand Minimization
A total of 8 Curcumin derivatives and 5 Allicin derivatives were used for study. In the Autodock all hydrogens were added to the ligands, Gasteiger charges were computed and added, and nonpolar H were merged; Autodock 4 atom types were assigned. It was ensured that total charge corresponds to tautomeric state. The torsion tree root & rotatable bonds were chosen as per requirement. The ligands were saved in the .pdbqt format. For the docking in Discovery Studio, the ligands were minimized using the Full minimization module. The ligands were minimized using the Smart minimizer algorithm with maximum steps of 2000, applying the CHARMm forcefield. The partial charge estimation was done by Momany-Rone parameters and the RMS gradient was set at 0.01. The minimized dataset was thus used for further analysis.
Docking Analysis of Receptor-Ligand interactions
The receptor active site cavity was identified by already existing ligand in the crystal structure complex of the main protease and RdRp protein. The ligands were docked by using 3 docking tools- Autodock, High throughput virtual screening algorithm that identifies hotspots (Libdock) followed by CHARMm based molecular dynamics scheme (CDocker). The best ligands obtained from all the docking protocols were chosen as top hits. The Libdock gives a positive binding affinity score; The Autodock and CDocker give a negative binding affinity score and the molecules having least binding affinity were selected as leads and potential drugs for repurposing that can act against SARS-CoV-2.
Autodock is an automated procedure for predicting the interaction of ligands with biomacromolecular targets. Autodock Vina uses a Simpler scoring function that allows a faster search method, and provides reproducible results for larger systems with upwards of 20 flexible bonds. Autodock calculations are performed in several steps: 1) preparation of coordinate files using AutoDockTools, 2) precalculation of atomic affinities using AutoGrid, 3) docking of ligands using Autodock Vina, and 4) analysis of results using AutoDockTools. After the AutoGrid is set up, a configuration file containing the receptor information, Ligand information, AutoGrid information and output path is prepared. The script is run on command prompt and after the docking is completed the docking results are analyzed in a suitable visualization tool like Discovery Studio. The Libdock docking program performs the following steps using a set of pre-generated ligand conformations and a receptor with a specified binding site: Libdock uses protein site features referred to as HotSpots. HotSpots consist of two types: polar and apolar. A polar Hotspot is preferred by a polar ligand atom (for example a hydrogen bond donor or acceptor) and an apolar Hotspot is preferred by an apolar atom (for example a carbon atom). The receptor Hotspot file is calculated prior to the docking procedure. The rigid ligand poses are placed into the active site and HotSpots are matched as triplets. The poses are pruned and a final optimization step is performed before the poses are scored. Ligand hydrogens, which are removed during the docking process, are added to the ligand poses. These hydrogens are not optimized, so they require further optimization to ensure that receptor-ligand hydrogen bonds are formed correctly. The high throughput screening of ligands was done using the FAST method of conformation. Energy threshold was set at 20 and maximum conformations were set at default value of 255. The number of hotspots was at the default value of 100. The top poses of each ligand were shortlisted.
CDOCKER is a grid-based molecular docking method that employs CHARMm. The receptor is held rigid while the ligands are allowed to flex during the refinement. For predocked ligands, prior knowledge of the binding site is not required. Random ligand conformations are generated from the initial ligand structure through high temperature molecular dynamics, followed by random rotations. The random conformations are refined by gridbased (GRID 1) simulated annealing and a final grid-based or full forcefield minimization. The following steps are included in the CDOCKER protocol:
• A set of ligand conformations are generated using hightemperature molecular dynamics with different random seeds.
• Random orientations of the conformations are produced by translating the center of the ligand to a specified location within the receptor active site, and performing a series of random rotations. A softened energy is calculated and the orientation is kept if the energy is less than a specified threshold. This process continues until either the desired number of low-energy orientations is found, or the maximum numbers of bad orientations have been tried.
• Each orientation is subjected to simulated annealing molecular dynamics. The temperature is heated up to a high temperature then cooled to the target temperature.
• A final minimization of the ligand in the rigid receptor using non-softened potential is performed.
• For each final pose, the CHARMm energy (interaction energy plus ligand strain) and the interaction energy alone are calculated. The poses are sorted by CHARMm energy and the top scoring (most negative, thus favorable to binding) poses are retained.
The simulation docking algorithm used the default parameters of generating top 10 hits. The dynamic steps were set at 1000 and the dynamic temperature was also considered to be 1000. The maximum orientations to refine were 10. The simulated annealing model was chosen for simulation procedure with heating steps set at 2000, heating target temp of 700, number of cooling steps were 5000 and cooling target temperature was 300. After CDocker, the top poses having good binding affinity and best receptor-ligand interactions from all the 3 docking programs are chosen as best molecules those have the potential to act against COVID-19.
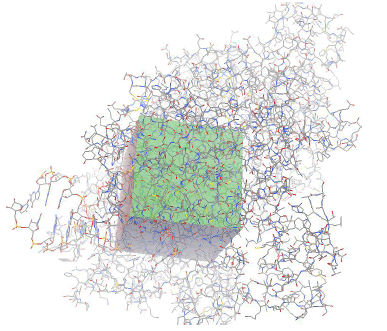
Target preparation and active site prediction The active site of the target protein was taken from the ligand present in complex with the protein the X-ray crystal structure. The following Table 1 depicts the sphere radius and coordinates of the active site.
Table 1: Active site of Main protease as well as RdRp.
| Protein | Main Protease | RNA dependent RNA Polymerase |
|---|---|---|
| (RdRp) | ||
| PDB id | 6LU7 | 7BV2 |
 |
 |
|
| Structure | ||
| Active Site | 24-26, 41, 49, 54, 140-145 and 163-192 | 295, 306, 310, 487, 545, 553, 619, 623, |
| residues | 642, 645, 646, 682, 691, 760,761, 800 | |
| XYZ coordinates | -10.7118, 12.4113, 68.8312 | 91.758007, 92.459430, 103.771683 |
| Active site radius | 13.807549 | 6.345 |
| Autogrid Map | 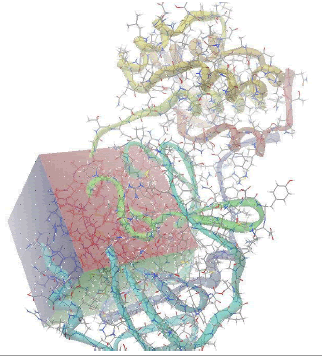 |
 |
Ligand minimization
The following Table 2 shows the minimization energy of the ligands of the Curcumin and Allicin derivatives.
Table 2: Minimization energy of Curcumin and Allicin derivatives.
| Curcumin derivatives | Minimization Energy | Allicin derivatives | Minimization Energy |
|---|---|---|---|
| Curcumin Sulphate | 9.51212 | Allicin | 20.3711 |
| Bisdemethoxycurcumin | -22.3049 | Diallyl disulphide | 15.4276 |
| Demethoxycurcumin | -20.5913 | S-Allyl cysteine | 2.60355 |
| Curcumin | -9.09157 | Ajoene | 21.2985 |
| Dihydrocurcumin glucuronide | 29.5117 | Diallyl trisulfide | 13.6661 |
| Hexahydrocurcumin | -7.2599 | ||
| Hexahydrocurcuminol | 3.65258 | ||
| Tetrahydrocurcumin | -11.9158 |
Curcumin docking with main protease
The docking of Curcumin derivatives was carried out by 3 docking tools- Autodock, Libdock and CDocker. The results of the same are depicted in the Table 3. The Libdock and Autodock results showed Hexahydrocurcumin and Hexahydrocurcuminol as the best docked molecule. The Autodock score for both the molecules was -6-6kcal/ mol and that of Libdock was 157.256 for Hexahydrocurcumin. Both the molecules were fitted inside the active site cavity with common Hbond interactions as Thr190, Phe140, Ser144 and Pi- Alkyl interactions as Cys145 and Met165. Both these amino acid molecules namely cys145 and Met165 were found to be involved in Pi-Alkyl interactions with majority of Curcumin derivatives indicating a strong pharmacophoric point at these aminoacids. Figure 6 depicts the interactions of Hexahydrocurcuminol with main protease by all 3 methods- Autodock, Libdock and Cdock along with the 3D view of protein cavity. Figure 7 shows interactions of Hexahydrocurcumin with main protease.
Table 3: Docking results of curcumin derivatives with main protease.
| Name of Molecule | Autodock Score | Libdock Score | CDocker Score | Interactions | |
|---|---|---|---|---|---|
| Curcumin derivatives | Hbond | Others | |||
| Curcumin Sulphate | -6.4 | 146.994 | 9.37408 | Glu166, Met165, Arg188, Asn142 | Cys145 |
| Bisdemethoxycurcumin | -6.5 | 103.823 | 22.8181 | Glu166, Asn142 | Cys145 |
| Demethoxycurcumin | -6.5 | 105.497 | 22.6971 | Met165 | |
| Curcumin | -6.3 | 90.4369 | 20.3708 | Gly143, Glu166, Asn142 | Cys145 |
| Dihydrocurcumin glucuronide | -6.1 | 140.327 | -1.16307 | Met165, Pro168 | Met165 |
| Hexahydrocurcumin | -6.6 | 157.256 | 33.5562 | Thr190, Phe140, Ser144 | Cys145, Met165 |
| Hexahydrocurcuminol | -6.6 | 138.225 | 30.6887 | His163, Thr190 | Cys145, Met165, His41 |
| Tetrahydrocurcumin | -6.3 | 139.386 | 25.9438 | Thr190, Ser144, Asn142, Leu141 | Cys145, Met165 |

Figure 6: Docking interactions of Hexahydrocurcuminol with main protease.

Figure 7: Docking interactions of Hexahydrocurcumin with main protease.
One of the important aminoacids involved in direct hydrogen bonding with almost all Curcumin derivatives was Glu166. Thr190, Ser144 and Asn142 were other important aminoacids directly involved in carbon hydrogen bond with the ligands Table 3 shows docking results of Curcumin derivatives with their interaction with amino acid residues.
Curcumin docking with RdRp
Although not previously predicted, the Sars-Cov nsp12 contains two metal-binding sites. The first is coordinated by His295, Cys301, Cys306, and Cys310. The second is in the fingers domain and is coordinated by Cys487, His642, Cys645, and Cys646. All eight of these metal-coordinating a.a. are highly conserved across the CoV family. Both of these metal-binding sites are distal to known active sites as well as protein–protein and protein–RNA interactions. Thus these ions were expected to be structural components of the folded protein rather than directly involved in enzymatic activity.
In the docking analysis of RdRp protein we could identify metal atoms such as Mg and POP bound to some of the atoms on the ligands. There were RNA-ligand, Metal atoms-ligand and proteinligand interactions found in the active site of the protein. The poly cysteine conserved domain was very well observed in the active site sometimes as directly interacting with the ligands or sometimes as van der Waals forces surrounding the ligands. Table 4 shows the docking results of Curcumin derivatives with RNA dependent RNA polymerase protein.
Table 4: Docking results of curcumin derivatives with RNA dependent RNA polymerase (RdRp).
| Name of Molecule | Autodock | Libdock | CDocker | Interactions | |||
|---|---|---|---|---|---|---|---|
| Score | Score | Score | |||||
| Curcumin derivatives | Hbond | Other | RNA | Others | |||
| Bonds | |||||||
| Curcumin Sulphate | -8.1 | 139.727 | 25.5717 | Asp623, | Arg555 | U20, | POP1003, |
| Asn691 | A11 | Mg1004 | |||||
| Bisdemethoxycurcumin | -7 | 101.116 | 33.0656 | Asn691, | U20 | Mg101 | |
| Arg555 | |||||||
| Demethoxycurcumin | -8.2 | 100.11 | 32.6066 | Asn691, Lys545 | U20 | POP1003 | |
| Curcumin | -8.5 | 88.6792 | 28.3405 | Asn691, Ser759 | Arg555 | U20 | POP1003 |
| Dihydrocurcumin glucuronide | -9.5 | 86.8381 | 8.90156 | Arg555, Ser682 | U20 | POP1003, Mg1004 | |
| Hexahydrocurcumin | -7.5 | 132.902 | 42.0269 | Arg553, Ser682 | U10, U20, A11 | ||
| Hexahydrocurcuminol | -7.6 | 135.599 | 38.9386 | Ser682, Asp760 | Arg555 | U10, U20 | POP1003, Mg101 |
| Tetrahydrocurcumin | -7.2 | 128.329 | 34.7815 | Asn691, Ser759 | U20 | ||
The best docked ligand was Dihydrocurcumin glucuronide found in Autodock with a score of -9.5 as well as in CDocker. From the Libdock results Curcumin sulphate was obtained as the best docked ligand with a score of 139.727. Aminoacid residues such as Asp623, Asn691, Arg555 and Ser682 were highly involved in the hydrogen bonding with most of the ligands Figure 8 depicts interactions of Dihydrocurcumin with RdRp protein. There were RNA interactions involved with U20, A11 and U10 atoms of the RNA. The POP1003 and Mg atoms were also directly bonded with most of the ligands giving stability to the structure from all sides. Figure 9 shows docking interactions of Curcumin sulphate with RdRp protein. The docking results of Curcumin derivatives are given in Table 4.

Figure 8: Docking interactions of Dihydrocurcumin glucuronide with RdRp protein.

Figure 9: Docking interactions of Curcumin sulphate with RdRp protein.
Allicin docking with main protease
Similar to Curcumin derivatives, Allicin derivatives were also docked with Main protease as well as RdRp protein, first with Autodock followed by Libdock and CDocker. The docking results of Allicin derivatives with Main protease are given in Table 5.
Table 5: Docking results of allicin derivatives with main protease.
| Name of Molecule | Autodock | Libdock | CDocker | Interactions | |
|---|---|---|---|---|---|
| Score | Score | Score | |||
| Allicin derivatives | Hbond | Others | |||
| Gly143, Ser144, | His41, Met49, Leu167, Met165, | ||||
| Allicin | -3.9 | 63.7364 | -3.9204 | Cys145 | Pro168, His163 |
| Met49, Met165, Pro168, Leu167, | |||||
| Diallyl disulphide | -3.1 | 58.0291 | -0.2782 | His172 | |
| S-Allyl cysteine | -3.1 | 71.2772 | 12.7073 | His41, Arg188, | |
| Leu141, His172, | |||||
| His163, Phe140, Glu166 | |||||
| Ajoene | -3.6 | 85.658 | -0.492665 | Thr190, His163 | His41, Met165, Leu167, Arg188, Pro168 |
| Diallyl trisulfide | -3.1 | 66.5068 | 1.82295 | Met49, Pro52, Met165, Pro168, Leu167, His41, Cys145 | |
From the Autodock and CDocker the best docked molecule was Allicin with a score of -3.9 for both Autodock and CDocker, followed by Ajoene with a best docked score of 85.658 for Libdock. The aminoacid residues such as Ser144, Cys145, Glu166, His41, Met165, Pro168 and Leu167 were involved in either H-bond or Pi-Alkyl interactions with the ligands. Figure 10 shows docking interactions of Allicin with main protease. Figure 11 shows Docking interactions of Ajoene with Main protease.

Figure 10: Docking interactions of Allicin with main protease.

Figure 11: Docking interactions of Ajoene with main protease.
Allicin docking with RdRp
The docking of Allicin and its derivatives was conducted first with Autodock, followed by Libdock and CDocker. Table 6 shows the docking results of Allicin and its derivatives with RdRp protein. Figure 12 shows the docking interactions of allicin with RdRp protein. Based on the Autodock and Cdock score, Allicin was found to be the best docked molecule with highest binding affinity of -4.6 and -2.066 for Autodock and CDock respectively. Second best molecule which was found to have good binding affinity was Ajoene and its score was found to be highest in the overall Libdock scores. Figure 13 shows Docking interactions of Ajoene with RdRp protein.
Table 6: Docking results of allicin derivatives with RNA dependent RNA polymerase (RdRp).
| Name of Molecule | Autodock Score | Libdock Score | CDocker Score | Interactions | |||
|---|---|---|---|---|---|---|---|
| Allicin derivatives | HBond | Other Bonds | RNA | Others | |||
| Allicin | -4.6 | 58.6118 | -2.06614 | Thr687, Asn691 | Cys622 | U10, A11, A19, A13, U20 | |
| Diallyl disulphide | -3.3 | 50.7507 | 2.18464 | Thr687 | Val557, Ala688, Ala547, Phe441, Ile548 | U10, U20, A11 | |
| S-Allyl cysteine | -4.2 | 67.3071 | 22.8974 | Ser759, Asn691, thr687, Ala688 | Val557, Cys622 | U20, U10, A11 | POP1003, Mg101 |
| Ajoene | -4.2 | 75.2643 | 1.62114 | Asn691, Ala688, Ser759 | Lys545, Arg555, Val557 | A11, U10, U20 | |
| Diallyl trisulfide | -3.3 | 55.9366 | 3.02276 | Asn691 | Cys622, Val557, Ala547, Phe441, Ile548 | U20, U10, A11 | |

Figure 12: Docking interactions of Allicin with RdRp protein.

Figure 13: Docking interactions of Ajoene with RdRp protein.
The molecules were found to have interactions with the aminoacids mostly through Pi-alkyl bonds. Here also POP1003 and Mg atom were found to be involved in the interactions again indicating their importance in the active site. The ligands were also found to have RNA interactions with U10, A11 and U20 atoms of the RNA either through direct Hydrogen bonds or through Pi-Alkyl interactions. Thus based on the overall combined results of Curcumin and Allicin derivatives the following Table 7 summarizes the top best molecules. Figures 14-17 shows combined interactions of all top molecules with protease and RdRp protein by all 3 methods Autodock, Libdock and Cdock.

Figure 14: Combined ligand interactions of Hexahydrocurcuminol and Hexahydrocurcumin from Autodock, Libdock and Cdock (Main protease).

Figure 15: Combined ligand interactions of Allicin and Ajoene from Autodock, Libdock and Cdock (Main protease).

Figure 16: Combined ligand interactions of Dihydrocurcumin glucuronide and Curcumin sulphate from Autodock, Libdock and Cdock (RdRp).

Figure 17: Combined ligand interactions of Allicin and Ajoene from Autodock, Libdock and Cdock (RdRp).
Table 7: Summary of docking results of curcumin and allicin derivatives with main protease and RNA dependent RNA polymerase (RdRp).
| Protein | Best Molecule | Best docked Score | Interactions |
|---|---|---|---|
| Main Protease | Hexahydrocurcumin | -6.6 | Thr190, Phe140, Ser144, Cys145, Met165 |
| Hexahydrocurcuminol | -6.6 | His163, Thr190, Cys145, Met165, His41 | |
| Allicin | -3.9 | Gly143, Ser144, Cys145, His41, Met49, Leu167, Met165, Pro168, His163 | |
| Ajoene | -3.6 | Thr190, His163, His41, Met165, Leu167, Arg188, Pro168 | |
| RdRp protein | Dihydrocurcumin glucuronide | -9.5 | Arg555, Ser682, U20, POP1003, Mg1004 |
| Curcumin sulphate | -8.5 | Asp623, Asn691, Arg555, U20, A11, POP1003, Mg1004 | |
| Allicin | -4.6 | Thr687, Asn691, Cys622, U10, A11, A19, A13, U20 | |
| Ajoene | -4.2 | Asn691, Ala688, Ser759, Lys545, Arg555, Val557, A11, U10, U20 |
Since the COVID-19 has become a global pandemic and affecting nearly all the countries worldwide, fast and effective ways to cure and prevent it has become an utmost necessity. Scientists all over the world are using different techniques such as in silico, in vitro and in vivo to find a cure for the disease. Here we have taken 2 naturally present compounds with established medicinal properties such as Curcumin and Allicin for testing their antiviral properties against SARS-CoV-2 proteins. We found compounds such as Hexahydrocurcumin, Hexahydrourcuminol, Dihydrocurcumin glucuronide and Curcumin sulphate showing good binding. Amongst the Allicin derivatives, Allicin and Ajoene showed good binding affinity for both Main protease as well as RdRp protein. Thus these molecules can be repurposed for their use as antivirals and can be studied for their in vitro and in vivo activity testing against COVID-19.
Authors reports no conflict of interest
On behalf of Amity Institute of Biotechnology, Amity University, Mumbai we thank Dassaults Systems for sponsoring BIOVIA license to support the challenging COVID-19 drug research.
Citation: Bastikar VA, Bastikar AV, Chhajed SS (2020) Understanding the Role of Natural Medicinal Compounds Such as Curcumin and Allicin against SARS-CoV-2 Proteins as Potential Treatment against COVID-19: An In silico Approach. J Proteomics Bioinform. 13:7. doi: 10.35248/0974-276X.1000510
Received: 17-Jun-2020 Accepted: 16-Jul-2020 Published: 23-Jul-2020 , DOI: 10.35248/0974-276X.1000510
Copyright: �© 2020 Bastikar VA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.