Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2017) Volume 6, Issue 3
ToxDelta is a new tool for the evaluation of the toxicity of chemicals based on a modified version of the fmcs_R package. Two structurally similar molecules share a maximum common substructure (MCS). In order to evaluate if two similar molecules have different effects, we focused our attention on the molecular fragments which are not in the MCS. These parts may increase or decrease the value of the property. We considered a variation of the MCS concept of efficient relevance in toxicity assessment where the rings of molecules must not be broken. To assess the toxicity of the target chemical, ToxDelta extracts the MCS and delineates the remaining fragments. Each of these moieties represents a difference between two molecules and its relevance in the toxicity assessment is evaluated against a knowledge-based list of active and inactive fragments. ToxDelta considers the dissimilarities of the molecules in a read across approach.
<Keywords: Read across; (Q)SAR, SAR; Maximum Common Substructure; Molecular graph; Toxicity assessment; Mutagenicity; Structural alert
The European REACH legislation for industrial chemicals promotes the use of alternative methods, and explicitly mentions and regulates the use of read across and quantitative structure-activity relationships (QSAR), jointly named non-testing methods (NTM). Often, one of the problems of QSAR models is their poor interpretability. Along with the assessment of the predictive power and statistical quality, the interpretability of the QSAR models is an important issue for the regulatory bodies. Furthermore, since read across is related to the concept of similarity, it is closer to evidence and apparently easier to be accepted, although similarity cannot be univocally defined. The European Chemicals Agency (ECHA) published a document with the purpose to communicate the framework applied within the agency to evaluate the assessment done with read across [1]. In 2014, it was reported that the most common and widely used NTM consisted in building categories and predicting properties by read across. Up to 75% of the analysed dossiers contained read across at least for one endpoint. The ECHA guidance on QSARs and grouping of chemicals introduces a flowchart [2] for the generation and use of non-testing data in the regulatory assessment of chemicals. This flowchart consists of a sequence of operations (eight steps), which starts with information collection and terminates with the final assessment exploiting the functionalities of a vast range of computational tools and databases. Depending on the chemical and property of interest these steps can be omitted or performed in a different order. In our new tool ToxDelta, we have addressed two of these steps: “Search for structural alerts” and “read across”. Even though read across has been used much more than QSAR for registrations, it has been studied much less than QSAR. There are many open issues on the use of this approach. Read across is typically subjective, and strongly relies on the individual expert, the expert’s background and experience, and is difficult to reproduce [3].
In order to solve the above mentioned problems of interpretation and to help the expert to get a documented, transparent and reproducible evaluation on the activity of the target compound, our group developed a new read across tool: ToxRead [3,4]. This tool assists experts in the evaluation of the biological activity/toxicity of compounds, offering known elements affecting the activity within the same picture.
Recently, we published the results of an exercise on read across. Participants made their assessment using the approach they preferred. The group of scientists who used ToxRead gave consistent assessment for the same chemical, while those who used other programs typically gave conflicting assessment [5]. This indicates that the subjectivity of the assessment may introduce a source of variability which may make read across an unreliable strategy without a proper reproducible scheme.
Generally, the programs assisting the expert in read across are based on similarity measuring software. Examples of these programs are ToxRead, the OECD QSAR toolbox [6], ToxMatch [7] and AMBIT [8]. VEGA, which is commonly used for QSAR, can be also used as a read across tool, as it shows the similar compounds and in many of its models also the alerts [3]. Similarity is basically measured on the basis of the chemical structure. In some cases additional toxicological considerations are added. These programs just show the most similar compound(s) to the target substance, and the user can decide the biological activity/toxicity of the target compound on the basis of the activities of the similar compounds used as source compounds. Furthermore, some of these programs (e.g. ToxRead) provide the value predicted by the software.
In combinatorial chemistry, the use of similarity and diversity methods addresses the similarity property principle. This principle states that structurally similar molecules have similar biological activity [9]. This statement is questioned by various experiences with contradictory results [10]. In fact, structural similarity does not always imply similarity in either activity [11] or descriptors [12]. Minor modifications can make active molecules to lose their activities completely and vice versa. Intrinsically, the similarity concept includes the fact that the two molecules are different. Thus, the expert should evaluate not only how similar the two substances are, but also whether the differences trigger an opposite behaviour.
Many similarity measuring methods have been proposed to quantify the similarity between chemical compounds especially in drug discovery research. One of the most famous methods is the study of substructure and superstructure relationships of the chemicals. Two molecules may share some common properties due to their common substructure. This search strategy does not provide any quantitative similarity measurement. Hence, it is a very knowledge-based approach in which every substructure used in a query needs to be well defined. Structural descriptor-based methods are another commonly used structural similarity searching approach in which the similarity of the chemical compounds can be quantified. Structure similarity search does not require an exact match and the search results are ranked by scores. One of the important structural-based search methods is fingerprint [13]. In this method, the chemical structure is disclosed in a highdimensional space. Many models for predicting biological activities are based on the similarity coefficient provided by such methods, such as neural networks [14], fuzzy adaptive least squares [15] and inductive logic programming [16]. Structural descriptor-based methods are computationally simple, but they are unable to identify local similarities between structures.
Maximum common substructure (MCS) is an encouraging approach for similarity searching and biological activities predictions in chemoinformatics. The MCS is a problem of graph matching that involves 2D or 3D chemical structures of two chemicals and identifies the largest substructure present in both molecule structures. The MCS-based methods have all the advantages of the substructure and superstructure -based methods and in addition does not need an exact match procedure. Compared to structural descriptor–based methods, MCS provides a similarity score for each comparison and can perform local similarity identification. MCS is a straightforward concept of determining similarities with a clear chemical meaning and is principally independent of the fingerprints. Several available MCS algorithms in the literature do not satisfy the graph representation of the chemical compounds. Barrow and Burstall in 1976 [17] used the MCS concept for the sub-graph isomorphism for the first time. After that, Cone et al. [18] introduced the use of MCS for similarity search for molecular comparison. The approach did not receive a notable consideration due to its complexity. Later, other MCS-based similarity search algorithms have been presented [19-21]. The concept of MCS in the molecule structures has been applied in different chemoinformatic concepts, such as classification models using the structural similarities [22], enrichment of chemical libraries [23] and clustering molecules with similar structural features [24]. The MCS search methods are mainly divided into “clique” [17,25] and “backtracking” [26,27]. The computational problem of finding all the largest complete sub-graph(s) (maximal clique) is called the clique problem. The clique problem is NP-complete, i.e., no polynomial time algorithm has been found to solve the general problem. However, many algorithms for computing cliques have been developed, both complete and approximate. The basic algorithm is due to Ullmann [28] who introduced backtracking to reduce the size of the search space. The MCS extraction algorithm of the FMCS R package is based on backtracking search method.
We considered a variation of the MCS concept of efficient relevance in toxicity assessment, where the rings of molecules must not be broken. We modified the MCS algorithm of the fmcs_R package [29] for finding the MCS between two given similar molecule graphs subject to this constraint. The similarity index is determined by the VEGA similarity indication which is described described by Floris et al. [30]. The new software, ToxDelta is a novel read across tool developed to identify and extract the differences between the target and the reference compounds for the further evaluation of the biological activity/toxicity of the target molecule. These differences are depicted as molecular substructures. Their possible role in amplifying or reducing the activity/toxicity of a compound is queried in an a priori prepared data set of molecular structural alerts (SA).
We added the constraint of keeping the aromatic or aliphatic rings present in the target or the reference molecule complete during the process of the MCS extraction. Indeed, this decision is made due to the important role that rings play in the mutagenicity and carcinogenicity SAs. For example, polycyclic aromatic hydrocarbons (PAHs) that are composed of multiple aromatic rings are a class of mutagens. In addition, PAHs are linked to skin, lung, bladder, liver, and stomach cancers in confirmed animal models. The increasing number of aromatic rings in PAHs helps the metabolic activation to reactivate diol epoxide intermediates and consequently their binding to DNA [31]. In addition, the mutagenicity of the aliphatic epoxides has been determined by the Ames test [32]. Historically, a very effective list of the SAs has been created and revised by Ashby in 1985 and 1988, respectively [33,34]. The Ashby’s well-known poly-carcinogen list contains aromatic nitro groups, aromatic azo groups, aromatic rings N-oxides, aromatic monoand di-alkylamino groups, aromatic amines and aliphatic and aromatic epoxides. The extended SAs list according to Kazius et al. [35] contains groups of specific aromatic nitro and amine, aliphatic halide, polycylic aromatic system and other SA with aromatic or aliphatic rings. Also, the Benigni’s [36] list includes an important number of forms of aromatic and aliphatic rings.
In our study, we combine a substructure identification tool with a tool for the assessment of the related fragments which are not in common, in order to evaluate the toxicity of the two chemical compounds under examination.
At present, ToxDelta performs only mutagenicity assessment of the chemical substances. The mutagenicity SAs collection of this tool is extracted from the Ames test results. Ames test is the gold standard for initial examination for detecting chemically induced gene mutations of new chemicals and drugs. Bruce Ames created the Ames assay in the 1970s [37]. The assay’s sensitivity towards many types of mutagens has been improved over the years [38,39]. Specific distinct mutations in the histidine and tryptophan synthetic pathways of Salmonella typhimurium and Escherichia coli have been created respectively, that result in the requirement for an exogenous supply of those amino acids for growth. Using genetically bacterial strains, the Ames test produces a high rate of inter-laboratory reproducibility (85%-90%) [40]. This assay has been proved to be the most predictive in vitro assay for rodent and human carcinogenicity [37,41]. Additionally, the Ames test results have been demonstrated to be in agreement with rodent carcinogenicity or in vivo genetic toxicity about 65% [42].
Database of active and inactive structural alerts
In a previous study, a very sophisticated collection of mutagenicity SAs has been created and implemented in ToxRead for the read across mutagenicity assessment [4]. This set of rules associated to bacterial mutagenicity has been identified and extracted by analyzing more than 6000 chemicals from different chemical classes. A set of rules related to both mutagenicity and lack of mutagenicity were found. These SAs have been sorted in a hierarchy of rules and used to identify the active or inactive mutagenic substructures present in the target compounds. The hierarchical order of the SAs makes it possible to identify first the exact rule that matches the target molecule and then other, more generic ones, which may match with the target molecule. Besides rules for mutagenicity and non-mutagenicity, the identified potential rules include exceptions and modulators of activity. These rules can be also used to predict mutagenicity concerning the influence of each SA found in the molecule. Accuracy and p-value are two statistical characterizations which are assigned to each SA of the mutagenicity list; these values show the accuracy of the SA based on the number of chemicals in the original training set containing the SA, and the prevalence of one of the categories: Mutagenic or non-mutagenic. These SAs are those implemented within ToxRead. In the case of the module for mutagenicity, there are about 800 SAs each with a high level of detail such as accuracy and statistical significance.
The MCS algorithm: ToxDelta advances ToxRead for it supports the reasoning based on the differences, as well as similarity of molecules. The degree of similarity and dissimilarity between pairs of molecules is computed from their structures. Molecular structures can be encoded in several computer formats which basically contain the topological information about the structure, as well as other chemical information such as atom charges, aromaticity, etc. Among several available formats, we relied on SMILES strings [43] and structure data formats (SDF).
The algorithm proposed in the fMCS_R package [44] performs MCS computation via a novel backtracking algorithm by incrementally computing a search tree of correspondences between nodes of the two graphs under investigation. Each node in this tree is a set of atom correspondences while leafs are the connected sub-graphs we are looking for; the deepest leafs are the MCSs.
The aromatic and non-aromatic rings as structural properties of molecules and their role in the biological activities of the molecules are important issues. Indeed, among the identified mutagenic and carcinogenic SA, aliphatic and aromatic rings play an important role. In this regard, an important number of forms of rings are established [36]. For this reason, we applied the constraint that the rings present in the input molecular graphs must be retained by the MCS by adding an additional check in the backtracking algorithm of the fmcs_R library, as opposed to the original R package (fmcs_R). In other words, we decided to maintain all the rings entire and not break these rings during the process of the MCS extraction. Our modification to the original code of this library consists mainly in adding a restriction during the process of the atom selection for the MCS. For each atom belonging to a ring, this restriction checks if the atom resides in an equivalent ring in the target and the reference molecule. Since the fmcs_R algorithm does not consider rings as such, it may break some rings, i.e. if it is necessary it selects only a subset of atoms in a ring. This leads to a significant loss of structural information and consequently the implication of the extracted MCS which is meant to be equal for both molecules may differ for each compound. We can finally extract the structural differences between the two compounds under investigation: we overlap each graph with the MCS and highlight all the sub-branches not in the MCS (Figure 1).

Figure 1: The MCS between two molecules is shown with bold lines and the other branches are the differences.
ToxDelta Implementation
The user can evaluate the evidences obtained by ToxDelta and make a decision regarding the toxicity of a compound under evaluation, in a weight of evidence approach. Figure 2 shows the flow chart of the implementation of the ToxDelta program for the mutagenicity endpoint. The new tool relies on ToxRead for the evaluation of the degree of similarity between similar compounds. The similarity algorithm has been described elsewhere [30]. A stand-alone version of ToxDelta is accessible on the VEGA home page (https://www.vegahub.eu/). ToxDelta will be implemented inside ToxRead and the dissimilar substructures will be computed between the target molecule and any source molecule selected by the user. ToxRead associates the most similar molecules present in its data base to the target molecule, pointing out the mutagenic (or non-mutagenic) fragment(s) as toxicity rules present in both the target and the similar chemical compounds. ToxRead identifies the mutagenic or non-mutagenic SAs in common between the target and the source chemicals. Thus, these SAs belong by definition to the MCS of the pair of compounds under investigation. At this point, the integration of ToxDelta inside ToxRead will allow further investigation of the pair of compounds, identifying the dissimilar moieties and providing the most similar SAs for each of them in the collection of the known SAs. To obtain a conceivable result, the structure of the target and the source molecules in the comparison need to be sufficiently similar. If the structures of the molecules compared by ToxDelta do not share a significant MCS, the dissimilarities may not be interpretable to an acceptable level. In other words, whenever the structures of two molecules are strongly dissimilar, the user may not expect a significant MCS. In this regard the VEGA chemical similarity index 30 is used as a screening before applying the MCS approach.

Figure 2: The flow chart of ToxDelta: The molecular similarity/dissimilarity structure analysis software for the mutagenicity endpoint.
Provided that the identified dissimilar fragment in the target molecule is an SA along with the assigned active or inactive toxicity effect information, there are three possible scenarios that may help the user to move in a certain direction for toxicity decision making. These three scenarios about the dissimilar fragment found in the target molecule are as following:
1. The SA is an active fragment with strong evidence which increases the effect;
2. The SA is an inactive fragment with strong evidence which decreases the effect;
3. The SA is a fragment without any relevant impact on the effect.
In case 1 and 2 the SA is more likely to modulate the effect of the whole molecule, while in case 3 the SA is a neutral fragment and does not have an impact on the modulation of the effect. Nevertheless, the software provides documentation on the SAs of case 3, which indicates the existence of a certain fragment with no impact on the effect. Documentation is an important factor in the acceptance of the read across results. This whole list of SAs is used by ToxDelta to assess whether the fragments resulting from the subtraction of the MCS from the molecule are associated to an increased or decreased or neutral effect.
The program returns as output all the possible MCSs of the same length (the number of the atoms is equal in all the MCSs) extracted from two molecules of interest. The user can choose one of the MCSs found and evaluate the dissimilarities calculated based on the chosen MCS. The different fragments present in both molecules, are the result of the subtraction of the MCS and the target or source molecules.
Evaluation of ToxDelta
In order to evaluate the new tool and to investigate its dissimilarity approach theory, we performed tens of in-house tests while studying and developing the proposed methodology. We selected two pairs of molecules with known mutagenicity (Ames test) experimental value as case studies, to show how our approach works and how it could be useful in the toxicity assessment. Even though there is no similarity threshold determined by this tool, for the molecules selected as case studies, we chose two pairs of molecules, case 1 and case 2, with a cut-off value of 0.7 for the VEGA similarity index [30]. The results provided by ToxDelta for the molecules with a small MCS may not have a significant interpretation. In both cases, we chose two compounds with different toxicity activity (one mutagenic and the other one non-mutagenic), as this scenario represents exactly the type of situation in which ToxDelta can provide useful insight. To check whether the structural differences between these molecules have a significant role in their toxicity or non-toxicity property, we assume that one of the molecules in each pair is the target molecule and the other one is the source molecule. We selected two pairs of derivatives from two relevant pharmaceutical classes: benzodiazepines and androstane derivatives. We chose diazepam, first came on the market as Valium, a benzodiazepine drug typically producing a calming effect. It is commonly used to treat anxiety, alcohol withdrawal syndrome, benzodiazepine withdrawal syndrome, muscle spasms, seizures, trouble sleeping, and restless legs syndrome. Flunitrazepam, known as Rohypnol, is a benzodiazepine derivative that can cause anterograde amnesia; its importation has been banned by the U.S. Government (https://chem.nlm.nih.gov/ chemidplus/name/flunitrazepam). The similarity VEGA index value between these two benzodiazepines is 0.87. Despite this, they exhibit different toxicological profiles: Indeed Diazepam is experimentally non-mutagenic while flunitrazepam is mutagenic. As second case study, we provided two androstane derivatives: mepitiostane and a structural analogue, cholestan-6-one, 3-bromo-, cyclic 1,2-ethanediyl mercaptole, S,S,S',S'-tetraoxide, (3-beta,5-alpha)-. Metpitiostane is an antineoplastic agent inhibiting the expansion of estrogen-stimulated cancers by a competitive inhibition mechanism for the estrogen receptor (https://pubchem.ncbi.nlm.nih.gov/compound/mepitiostane#section=Pharmacology-and-Biochemistry). The similarity index value between these two chemicals is 0.77. We processed the selected molecules using ToxDelta to explain the discrepancy between mutagenic activities for each pair. The results of the ToxDelta tool are discussed in the results section.
The new ToxDelta software uses the structures of the two chemicals to be compared as input. The two substances are introduced as SMILES [43]. The MCS is the common part present in both molecules and it is shown (Table 1). This MCS is usually a large part of the molecules to be assessed. Indeed, the application of ToxDelta is useful for substances that are structurally similar. The MCS typically, even if implicitly, represents the driving force in the read across procedure. This is the logical process which identifies the analogies among substances. In this scheme, ToxDelta does not contradict but complements the read across conceptual strategy. The risk of the read across strategy is to miss the differences between two molecules. The similarity should not erase the possible opposed behaviour of the two similar compounds. But how to avoid the error of ignoring factors which may provoke opposite behaviour? ToxDelta wants to address this issue. It carefully identifies the differences and the related toxicological meaning. The theoretical basis is closely related to the SA paradigm. Thus, ToxDelta complements the ToxRead software, which exploits all the SAs of the target compound. Beyond this global assessment, done by ToxRead, it is useful to apply ToxDelta for a closer look at the two substances (i.e. the target and the reference compounds), in particular, when they may have opposite toxicological properties. Indeed, it should be reminded that ToxRead predicts the toxicological property of the target compound, and thus the predicted value of the target compound may be the opposite of the experimental value of the similar compound.
| Molecules | MCS* | Dissimilar fragments | ||
|---|---|---|---|---|
| Case study 1 | Target | 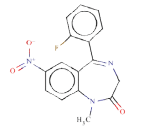 |
 |
 |
| Source | 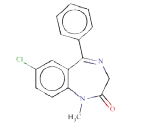 |
 |
||
| Case study 2 | Target | 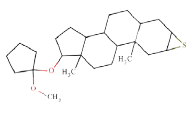 |
 |
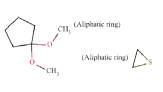 |
| Source | 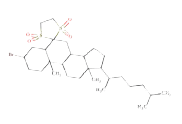 |
 |
* Maximum common substructure
Table 1: The two case studies: Case study 1) target molecule: Diazepam, source molecule: Flunitrazepam; Case study 2) target molecule: cholestan-6-one, 3-bromo-, cyclic 1,2-ethanediyl mercaptole, S,S,S',S'-tetraoxide, (3-beta,5-alpha)-, source molecule: mepitiostane and the results of ToxDelta: Maximum common substructure and dissimilar fragments.
Case study 1: Benzodiazepine derivatives
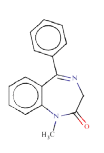
Source molecule 1: Diazepam
Systematic name: 1-methyl-5-phenyl-7-chloro-1,3-dihydro-2H- 1,4-benzodiazepin-2-one
SMILES: O=C1N(c3ccc(cc3(C(=NC1)c2ccccc2))Cl)C
Experimental activity: Non-mutagenic in Ames test [45]
CAS number: 439-14-5
Target molecule 1: Flunitrazepam
Systematic name: 1,3-dihydro-5-(o-fluorophenyl)-1-methyl-7- nitro-2H-1,4-benzodiazepin-2-one
SMILES: c12C(=NCC(=O)N(c1ccc(c2)[N+](=O)[O-])C)c1c(cccc1)F
Experimental activity: Mutagenic in Ames test [45]
CAS number: 1622-62-4
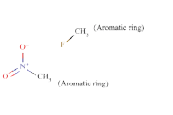
ToxDelta identifies “7-chloro-1-methyl-5-phenyl-2,3-dihydro- 1H-1,4-benzodiazepin-2-one” as MCS shared by these two chemicals (Table 1). ToxDelta also extracts three fragments of dissimilarities: the nitro group, the fluorine and chlorine atoms, each linked to an aromatic carbon. Diazepam lacks the first two fragments, which are present in Flunitrazepam. The nitroaromatic moiety matches two ToxRead SAs for mutagenicity both referring to the generic nitroaromatic ring; the Benigni–Bossa alert does not include chemicals with orthodistribution and with a sulphonic group on the nitroaromatic ring. This leads to a slight difference in the accuracies of these fragments, which are respectively 85% and 87%. ToxDelta identifies also the fluorine and chlorine atoms linked to aromatic carbons as dissimilarity fragments between the two molecules. These moieties do not match any rule for Ames mutagenicity included in the ToxRead “libraries” of SAs [3,4]. As a conclusion, ToxDelta immediately reports as a key difference the presence of the nitroaromatic fragment, which is at the basis of the different mutagenicity value of the two substances.
Case Study 2: Androstane derivatives
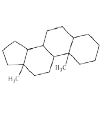
Source molecule 2: cholestan-6-one, 3-bromo-, cyclic 1,2-ethanediyl mercaptole, S,S,S',S'-tetraoxide, (3-beta,5-alpha)-
Systematic name: Cholestan-6-one, 3-bromo-, cyclic 1,2-ethanediyl mercaptole, S,S,S',S'-tetraoxide, (3-beta,5-alpha)-
SMILES: O=S5(=O)(CCS(=O)(=O)C35(CC1C4CCC(C(C) CCCC(C)C)C4(C)(CCC1C2(C)(CCC(CC23)Br)))
CAS number: 133331-34-7
Experimental activity: Mutagenic in Ames test [45]
Target molecule 2: mepitiostane
Systematic name: 5-alpha-Androstane, 2-alpha,3-alpha-epithio-17- beta-(1-methoxycyclopentyloxy)-
SMILES: O(C)C6(OC2CCC3C4CCC1CC5C(CC1(C) C4(CCC23(C)))S5)(CCCC6)
CAS number: 21362-69-6
Experimental activity: Non-mutagenic in Ames test [45]
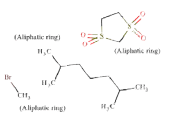
ToxDelta identifies the androstane tetracyclic system as MCS shared by these two chemicals and extracts five fragments of dissimilarity (Table 1). Three of these are aliphatic rings: the thiirane, 1,1-dimethoxycyclopentane, and 1,3-Dithiolane 1,1,3,3-tetraoxide rings and two are aliphatic chains: the 2-methylheptyl group and a bromine atom, both linked to an aliphatic carbon ring. The cyclic moieties and the alkyl carbon chain do not match any rule potentially responsible for mutagenic/non-mutagenic activity listed in the ToxRead software. Conversely, the bromine atom linked to an aliphatic carbon ring corresponds to two ToxRead SAs both referring to bromo-/haloethyl moieties with different levels of specificity and a prevalence of mutagenic activity of 71% and 67%, respectively. These rules, which are present in the source molecule but not in the target chemical, give a first indication of different toxicological profiles for these chemicals.
Evaluating the two case studies, it is important to notice that sometimes the identified dissimilar fragment is not an entire SA. In many cases the fragment of dissimilarity is a fraction of the whole rule (an already existing rule in the rule set) and the rest of the SA appears in the MCS. This issue is completely solved by the ToxRead software. In fact, the dissimilarities examination of ToxDelta takes place after the visualization of the results of ToxRead, when the user has already observed all the existing SAs that are matched with the target molecule and are in common between the target molecule and a set of structurally similar molecules.
ToxDelta is a new tool for read across concept not aimed at substituting other tools, but to complement them. It has been designed to match certain features of ToxRead, but it can also be used alone. It is important to underline that ToxDelta addresses differences between two molecules, and per se it does not address the overall toxic property of the molecule, while this aim may be accomplished by other tools, like ToxRead, covering the assessment of the target molecule. The main advantage of ToxDelta to the other read across programs is its focus on dissimilarities in addition to the similarities and the resembling properties between structurally similar compounds. It exploits the adverse effects that these dissimilar fragments may trigger in the biological activities or properties of the chemical substances.
ToxDelta provides a further insight by analysing the modulations of the effects which are expected in relation to the presence of the additional fragments in one of the two molecules under evaluation. Compared to other tools for read across, ToxDelta is more “local”, and this fact makes it an ideal tool to evaluate the effect of the metabolites and the impurities related to a compound having at hand the experimental values for the parent compound. Two important fields in which this issue can be applied are impurities in pharmaceutics and pesticides. The Food and Drug Administration (FDA) has provided a guideline for industry about the mutagenicity of the pharmaceutical impurities [46] that describes a practical framework for identification and control of the identified mutagenic impurities in order to limit potential carcinogenic risk. Another appropriate field of application for this tool is in pesticides assessment. The European Food Safety Authority (EFSA) has addressed the possible use of in silico methods for the evaluation of the effects of metabolites of pesticides [47]. ToxDelta may represent an ideal tool for pesticides, biocides and pharmaceutical compounds; because in these cases the experimental property values of the parent compound is requested by the relative regulations and ToxDelta can provide this information. Thus, ToxDelta may be particularly useful in those cases where data for the parent compound are available, and the user is interested not in the absolute effect of the related compound, but the possible increase of effect in an impurity product. For instance, if the toxicity level of the impurities is similar to the parent compound, this fact does not affect the way the substance with the impurity should be handled and regulated. Conversely, if the impurity represents an increased hazard, this may be a serious issue. To overcome this kind of problems, local tools that deal with measuring the relative increase or decrease of the effects are probably more accurate than absolute de novo predictions.
ToxDelta aims to address an important issue associated with read across. Although the use of read across approaches is widespread, the acceptance of the dossiers using read across is not straightforward. Detailed documentation has to be provided by the expert. One of the main sources of scepticism on the assessment of read across is that there are two (or more) substances under consideration, the target compound, lacking of data, and the reference compound, which is assumed to represent the properties of the target compound. So far the existing software for read across have focused on the assessment of similarity between the target and the source compounds, with the idea that the higher the similarity is, the higher is the likelihood that the properties of the two compounds will be similar. However, authorities often argue that even minor modifications of the chemical structure may provoke a dramatic change in the property value. To complement the existing software addressing similarity, we focused our attention on the differences between two compounds, introducing ToxDelta.
It is noticeable that unlike other read across programs, the SAs within ToxRead and ToxDelta do not exclusively contain active fragments, but also inactive fragments. This advantage allows exploring positive and negative modulations of the effect, and recognizing whether any relevant impact is expected. These SAs are associated to statistical characterizations, based on the number of chemicals containing the fragment, and on the prevalence of one of the categories: toxic or non-toxic. As a result, the user has both, the evidence that a certain fragment is associated to a certain effect and the statistics related to the prevalence of active or inactive compounds containing that SA. ToxRead provides all the data available on mutagenicity and BCF endpoints, and enables the user to access the available knowledge in a read across approach. This software with its genuine graphical user interface organizes different groups of similar molecules and allows the user to move in different levels of reasoning. ToxDelta nicely complements ToxRead, offering additional focus on all the fragments which may affect the toxicity.
Currently, a beta version of ToxDelta is freely available on the VEGA platform (https://www.vegahub.eu/) and the toxicity endpoint for which this tool can be used is mutagenicity. Other endpoints will be added to the software in the next future.
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Azadi Golbamaki and Alessio Mauro Franchi contributed equally.
This research was supported by the PROSIL project (LIFE12 ENV/IT/000154) and the EU-TOXRISK project. The EU-ToxRisk project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 681002.