Journal of Glycobiology
Open Access
ISSN: 2168-958X
ISSN: 2168-958X
Review Article - (2013) Volume 2, Issue 1
Uncomplicated pregnancies represent a hypercoagulable state. However, placental thrombosis is rare in these pregnancies which suggest that thrombin generation must be tightly regulated. In contrast, pregnancy disorders such as pre-eclampsia (PE) demonstrate an exaggerated increase in pro-coagulant activity contributing to the thrombotic lesions observed in the uteroplacental circulation of these pregnancies. Therefore, the tight haemostatic regulation observed in uncomplicated pregnancies is disrupted in PE-affected pregnancies.
Glycosaminoglycans (GAGs) are abundantly expressed within the placenta. GAGs do not exist in isolation, but are predominately bound to proteins known as proteoglycans (PGs). PGs have important anticoagulant, antiinflammatory and pro-angiogenic properties. Placentae from pregnancies complicated by PE demonstrate a reduction in PG expression. This may be a plausible explanation for the increase in thrombosis observed in this pregnancy complication.
Heparin is a well known pharmacological GAG with biochemical structure similar to the endogenous GAG, heparan sulphate (HS). Recent clinical studies have suggested that antenatal heparin therapy may reduce the likelihood of developing PE and fetal growth restriction (FGR). However, the mechanism by which heparin acts to reduce the likelihood of such pregnancy complications is very poorly understood and is necessary in order to improve the efficacy of this drug before it is to be used as a standard treatment.
Keywords: Glycosaminoglycans; Pre-eclampsia; Heparin
Pre-eclampsia (PE) is a serious pregnancy complication which affects 3-5% of pregnant women and contributes to over 60,000 maternal deaths worldwide per year [1]. PE is clinically defined as maternal hypertension (systolic BP ≥140 mmHg or diastolic BP ≥ 90 mmHg) and proteinuria (≥ 300 mg in a 24 h urine specimen) occurring after 20 weeks gestation [2]. Currently, there is no treatment for PE and management is reliant upon appropriate detection and timely delivery which may result in delivery of a very premature infant. This has long term repercussions for both mother and the baby. Mothers who experience PE have an increased rate of cardiovascular disease and premature death [3].
To date, the precise pathogenesis of PE is yet to be elucidated however, delivery of the placenta appears essential in aiding the resolution of this condition. This suggests that the fetus and therefore the placenta is a significant contributor to the pathogenesis of PE [1].
The predominant cell type in the human placenta is the trophoblast cell, which is particularly important in the regulation and development of a healthy placenta (Figure 1) [4]. Trophoblast cells differentiate into villous syncytiotrophoblasts, extravillous anchoring trophoblast cell columns or invasive, migratory intermediate trophoblast cells [5]. The villous trophoblast cells proliferate, differentiate and fuse to form the syncytiotrophoblast, a large multinucleate mass bathed in maternal blood. This syncytiotrophoblast layer provides an interface for the exchange of nutrients and waste between the mother and fetus, as well as being involved in growth factor and hormone production essential for maintaining pregnancy [6]. In order to ensure successful anchorage of the placenta to the uterus, a population of trophoblast cells proliferate to form an anchoring trophoblast cell column to facilitate the attachment of the placenta to the endometrium [7]. The extravillous cytotrophoblast cells are the invasive, migratory trophoblast cells. They travel into the decidua, surround and interact with the uterine spiral arterioles, replacing the smooth muscle cells with placental endothelial cells in order to remodel the spiral arteries [8]. This invasion and subsequent remodelling is crucial for the growing fetus as it creates a low resistance vessel to meet the increased demand in blood flow.
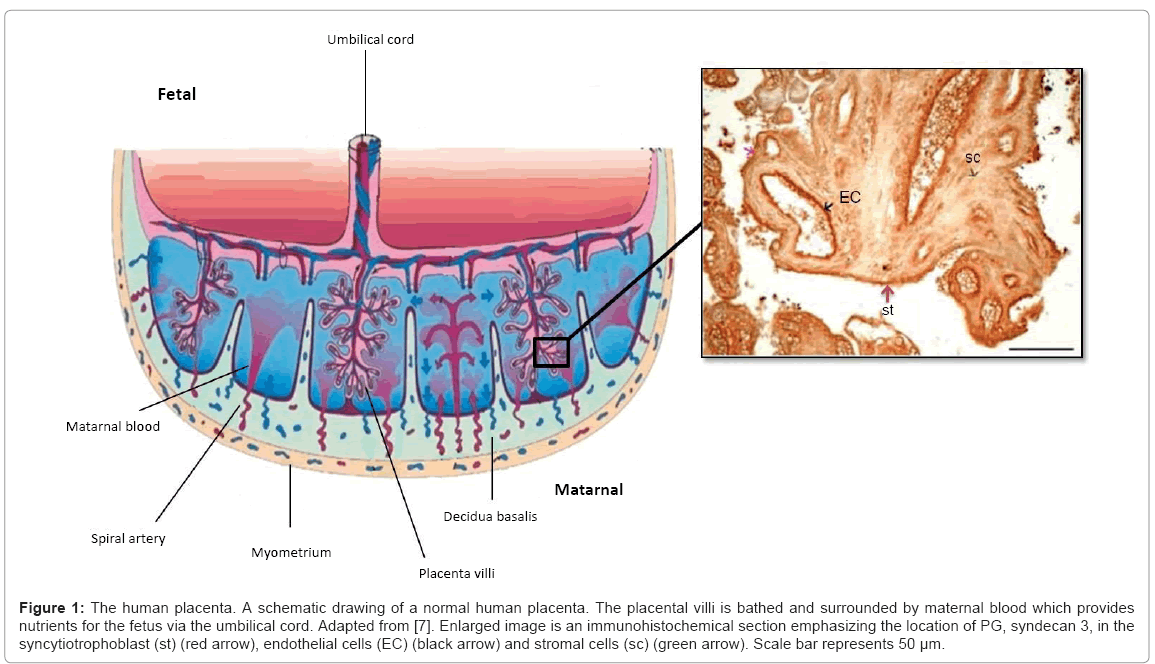
Figure 1: The human placenta. A schematic drawing of a normal human placenta. The placental villi is bathed and surrounded by maternal blood which provides nutrients for the fetus via the umbilical cord. Adapted from [7]. Enlarged image is an immunohistochemical section emphasizing the location of PG, syndecan 3, in the syncytiotrophoblast (st) (red arrow), endothelial cells (EC) (black arrow) and stromal cells (sc) (green arrow). Scale bar represents 50 μm.
Adequate fetal nutrition is crucial for normal fetal growth. The supply of nutrients can be affected by numerous factors including maternal diet, the quality of placentation and the efficiency of placental nutrient transfer [9].
Haemostasis represents equilibrium between the coagulation and fibrinolytic systems which ensures proper blood flow and clotting when necessary. It has been well established that uncomplicated pregnancies represent a hypercoagulable state [10]. Pregnancy is associated with an increase in the production of pro-coagulant factors (factors V, VII, VIII, IX, X, von Willibrand factor (vWf), thrombin), a decrease in circulating anticoagulant factors (proteins S) and a reduction in fibrinolytic activity to ensure blood loss during parturition is well regulated [11-14].
The significant increase of soluble fibrin by third trimester contributes to the increased thrombin generation observed in normal pregnancies [15]. It is important to note that fibrin formation is an important process for normal implantation of the placenta, in particular at sites of trophoblast invasion to allow expansion of the basal plate [16]. However, the haemostatic changes observed in uncomplicated pregnancies appear to be exaggerated in PE pregnancies. For instance, uncomplicated pregnancies show evidence of a small insignificant amount of fibrin deposition in the uteroplacental vasculature, but in PE there is excessive fibrin deposition in the spiral arterioles thus disrupting uteroplacental blood flow [17,18]. Higgins et al. also demonstrated that activation of the coagulation system is more marked in the uteroplacental circulation than in the systemic circulation suggesting that local factors may regulate haemostasis in the uteroplacental circulation [19].
The presence of thrombin is an indication of the activation of the coagulation cascade. Thrombin plays an important role in coagulation by converting fibrinogen to fibrin, activating platelets and other crucial clotting factors (factor V and VII) [19]. This process must be tightly regulated to prevent placental thrombosis or clotting within the maternal-fetal unit [20,21]. Therefore, in order to prevent increased thrombosis in placental vessels during uncomplicated pregnancies, tight regulation of thrombin inhibitors and anticoagulants is vital. Antithrombin and heparin cofactor II (HCII) are the major thrombin inhibitors in maternal and fetal plasma [22]. Evidence suggests that PE is associated with an imbalance in this haemostatic control [12,19].
In uncomplicated human pregnancies, maternal and fetal interactions create a mild systemic inflammatory state. However, in PE, there is an exaggeration of this inflammatory response [23,24]. Furthermore, fibrinogen production is known to be increased in inflammation which is further associated with thrombotic disease [25]. This may account for the increase in fibrinogen observed in PE pregnancies and the presence of thrombotic lesions within these placenta [26]. Inflammatory mediators such as tumour necrosis factor, CD40 and endotoxin are all inducers of tissue factor which may further facilitate the coagulation response [25] in these pregnancies. In addition, protein C, a naturally circulating anticoagulant is an important regulator of inflammatory pathways. A reduction in protein C activity has been reported in PE pregnancies [27] which may contribute to the increase in coagulation and inflammatory responses [28], thereby further exacerbating this condition.
Angiogenic factors also play important roles in regulating the development of placental vasculature. The expression of anti-angiogenic growth factors, soluble fms-like tyrosine kinase 1 (sFLT1), transforming growth factor β (TGF-β) and endoglin are upregulated in pregnancies affected with PE while pro-angiogenic growth factors such as vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) are downregulated in these pregnancies, and may account for the inadequate cytotrophoblast invasion, endothelial dysfunction and impaired placental angiogenesis that is commonly associated with PE [1,29,30].
Glycosaminoglycans (GAGs) are linear polysaccharides consisting of repeating disaccharide units [31]. GAGs are linked to a core protein and together are referred to as proteoglycans (PGs). There are four types of structurally distinct GAG chains, chondroitin sulphate (CS), dermatan sulphate (DS), heparan sulphate (HS) and hyaluronan (HA). HA is structurally the simplest GAG but is the only GAG that is not covalently linked to a core protein [32,33]. GAGs participate in a wide variety of biological functions [34].
PGs were initially thought to be important structural molecules; however emerging evidence has highlighted their importance in the regulation of cellular function, angiogenesis, inflammation, interaction with a number of growth factors and anticoagulation [35-37].
PGs are expressed abundantly within the human placenta (Table 1) and contain three major types of GAGs: HS, and CS or DS [38]. HS binds readily to the naturally circulating anticoagulant antithrombin through a unique pentasaccharide sequence found on one-third of the HS chain which subsequently induces a structural change in antithrombin to increase the rate at which antithrombin binds to thrombin [39]. DS chains binds to HCII through a charged sequence to inhibit thrombin production [40]. Together, these cell membranes bound GAGs contribute to localised thrombin regulation.
| Proteoglycans | GAG side chain | Localisation within the placenta | Function | Reference |
|---|---|---|---|---|
| Syndecan 1 | HS | Syncytiotrophoblast | Interact with numerous growth factors, wound healing, cell proliferation | [64,66,106,107] |
| Syndecan 2 | HS | Endothelial cells, smooth muscle cells | Regulating haemostasis, interact with growth factors i.e.: VEGF, role in cell migration | [64,67,69,71,108] |
| Syndecan 3 | HS | Endothelial cells, syncytiotrophoblast | Interact with growth factors, cytokines, acts as a co receptor | [64,109] |
| Syndecan 4 | HS | Fetal vessels, syncytiotrophoblast | Cell signalling, cell adhesion, migration | [64,110,111] |
| Biglycan | CS/DS | Endothelial cells | Matrix remodelling | [92,112] |
| Glypican 1 | HS | Syncytiotrophoblast | Cell adhesion, migration, cell signalling, growth factor interaction | [62,113] |
| Glypican 3 | HS | Syncytiotrophoblast | Implicated in thrombin generation, cell signalling, | [75,114] |
| Perlecan | HS | Syncytiotrophoblast | Facilitates trophoblast invasion, angiogenesis, wound healing | [64,67] |
| Decorin | CS/DS | Endothelial cells | Involved in cell proliferation, migration, invasion, angiogenesis | [93,115-117] |
Table 1: The nine PGs localised within placenta. Localisation of the nine PGs and their respective GAGs and their known functional roles within the placenta.
GAGs are also involved in other important biological functions such as inflammation and angiogenesis. During the inflammatory process, the structure and localisation of GAGs alters. GAGs are found in the wound fluid following an injury and in addition to its inflammatory activation, the GAGs go on to activate a range of growth factors [41,42]. Furthermore, inflammatory cytokines IL-6 and IL-10 are shown to induce GAG synthesis in endothelial cells which, in turn, lead to changes in angiogenesis resulting in increased vascularity [43]. Therefore, investigating the role and functions of PGs and GAGs within the placenta may provide an insight into the biological basis of placental mediated disorders such as PE.
Heparan sulphate proteoglycans (HSPGs) are found predominately on the cell surface and in the extracellular matrix (ECM) of mammalian cells [44]. Their location allows these PGs to interact with numerous growth factors, cytokines and cell signalling pathways [45]. HSPGs can either act as a receptor or co-receptor for various ligands. For example, HS acts as a co-receptor for numerous growth factors including fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) and is responsible for their activation leading to a number of cellular responses required for organogenesis, pattern formation and angiogenesis during development [46-48]. HSPGs also interact with a number of extracellular ligands and receptors which subsequently activate signalling pathways including; rat sarcoma-rapidly accelerated fibrosarcoma -mitogen activated protein kinases (Ras-Raf-MAPK) which is responsible for numerous physiological processes including cell cycle regulation, cell proliferation, survival and apoptosis [49] and also interacts with the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway which is implicated in organ development including cell survival, differentiation and proliferation [50].
The biosynthesis of HS involves three primary stages, chain initiation, polymerization and modification. Chain initiation involves the linkage of tetrasaccharide molecules onto serine residues in the core protein. Polymerization then takes place following the addition of an N-acetylglucosamine residue with alternating addition of glucuronic acid and N-acetylglucosamine residues [51]. Following expansion of the chain modifying enzymes such as NDST (n-deacetylase/n-sulfotransferase), C5-epimersase and 2-O-, 6-O- and 3-O- sulfotransferases add sulphate groups to specific locations of the chain [51]. This is in contrast to CS and DS biosynthesis where sulphation primarily occurs during chain polymerization [52].
The importance of individual modification enzymes such as C5-epimerase, 2-O- or 3-O sulfotransferases is highlighted in mice and flies that lack these enzymes as they experience early embryonic patterning defects and even perinatal death [53-55]. In addition, heparan sulphate 6-O sulfotransferase 1 (HS6ST-1) deficient mice are shown to exhibit aberrant angiogenesis in the placental labyrinth microvessels which suggests that 6-O-sulphate in HS may play a critical role in development [56,57]. Warda et al. demonstrated significant reductions in O-sulfotransferases in PE-affected placentae which also correlated with structural GAG changes, suggesting this altered GAG structure may alter the normal GAG functions, including regulation of placental haemostasis, thereby contributing to the pathogenesis of PE [58]. The three major classes of HSPGs are the syndecans, glypicans and the ECM class which includes, agrin, collagen XVIII and perlecan [59].
Syndecans are a family of four transmembrane proteins identified as syndecan 1, 2, 3 and 4 and are abundantly expressed in the human placenta [60-62]. Syndecans are comprised of HS, CS or DS chains but predominately HS side chains which allow them to interact with a number of growth factors that are involved in vascular development and repair [63].
Syndecan 1 is predominantly localised to the syncytiotrophoblast in term placentae [64]. Syndecan 1 has diverse roles including: acting as a low-affinity cell surface receptor for basic fibroblast growth factor which affects both cellular growth and angiogenesis [65], and regulation of cell proliferation [66]. Syndecan 1 mRNA and protein expression is significantly reduced in both PE [67] and FGR compared to uncomplicated pregnancies [64,68]. This suggests that syndecan 1 may play an important role in altering growth factor interaction and angiogenesis within the placentae.
Syndecan 2 is localised to the endothelial and smooth muscle cells of the fetal vessels in the placenta [64]. The location of syndecan 2 allows it to readily interact with a number of plasma proteins such as, antithrombin, lipoprotein lipase, as well as various growth factors and chemokines [69]. As a result of the close proximity syndecan 2 has to fetal blood it is plausible that the HS chains can easily come into contact with these plasma proteins to prevent local clot formation. Syndecan 2 also appears to have an important role in angiogenesis [70]. This is especially highlighted in syndecan 2 knockout mice which demonstrate reduced endothelial cell migration and capillary tube formation within the mouse brain and defective angiogenic signalling in zebrafish [71,72].
Pregnancies affected by both PE and FGR are shown to exhibit decreased syndecan 2 expression at both the mRNA and protein level [64,67]. Therefore, it is plausible that as a result of decreased syndecan 2 expressions there is reduced abundance of HS available to catalyse thrombin inhibitors in the fetal circulation which may result in a reduction in blood flow, commonly associated with PE and FGR [73].
Syndecan 3 and syndecan 4 are also expressed within placental tissues but in contrast to syndecan 1 and 2, no significant differences were observed in PE and FGR [64,67]. Syndecan 4 knockout mice demonstrate degeneration of the fetal vessels leading to a FGR-like phenotype [74].
Glypicans are a family of six HSPGs linked to the cell surface through a glycosylphosphatidyl anchor [44,59]. Glypicans 1 and 3 are reported to be abundantly expressed in both the syncytiotrophoblast and the villous cytotrophoblast cells of the human placenta [62,75]. Chui et al. demonstrated a highly significant decrease in glypican 1 and 3 mRNA and protein expression in human pregnancies complicated by PE compared to controls [67]. Glypicans bind to a number of growth factors and extracellular matrix (ECM) molecules including the tissue factor pathway inhibitor-2 (TFPI-2). TFPI-2 inhibits the tissue factor-factor VIIa complex that is normally formed to activate factor X which allows the conversion of prothrombin to thrombin. Therefore it is plausible that when glypican binds to TFPI-2 this would impair factor Xa activity and subsequently reduce thrombin generation [76]. Hence, the reductions in placental glypican 1 and/or 3 mRNA and protein expression observed in PE-affected pregnancies could contribute to the increase in local uteroplacental thrombin generation observed with PE [67] consistent with the findings of Higgins et al. [19].
The ECM PGs, which include perlecan, are a family of large PGs. Perlecan comprise five domains and is predominately expressed in the basement membrane and ECM of tissues. It is speculated to have an evolutionary role in nutrient metabolism, adhesion and angiogenesis [36,59,77].
Perlecan mRNA and protein expression is significantly decreased in pregnancies complicated by PE compared to their respective controls [67]. Perlecan binds with high affinity to Heparan-sulphate Interacting Protein (HIP), an important protein during trophoblast invasion and blood coagulation [78]. HIP is abundantly expressed in human cytotrophoblast cells and its expression is reduced in PE-affected pregnancies [79]. Thus, it is plausible that the reduced perlecan expression observed in PE-affected pregnancies may reflect the reduced availability of HS interacting with HIP, leading to inadequate trophoblast invasion. Likewise, reduced availability of HS may contribute to the coagulation disturbances which are commonly observed in PE-affected pregnancies [67].
Heparanases are endoglycosidases that are responsible for cleaving the HS GAGs from the core protein and degrading them to smaller fragments [80]. There are a number of heparanase proteins including heparanase 1 and 2, however heparanase 1 is the major protein responsible for degrading the HS side chains and is shown to be expressed mainly within the placenta [81]. Heparanases are reported to be pro-inflammatory as they are found to be most active at an acidic pH which is the case during inflammation [82]. Heparanase also exhibits pro-angiogenic properties by releasing heparin binding angiogenic growth factors such as FGF 2 and VEGF into the ECM [81,83,84].
Heparan-sulphate interacting protein (HIP) antagonizes HS degradation by heparanase by competing for the same binding site in the GAG chain which may account for the abundance of heparanase observed in normal placental tissue without any adverse outcomes [85,86]. As HIP expression is decreased in PE-affected pregnancies it is plausible that an imbalance in heparanase/HIP regulation could account for the increase inflammatory and altered angiogenic response observed in these pregnancies. A plausible treatment option for prevention of PE is pharmacologic heparins. Low molecular weight heparins and chemically modified heparin devoid of anticoagulant activity has been shown to inhibit degradation of HS in the ECM although heparanase inhibition was dependent on the degree of sulfation of the heparin molecule, and the position of the sulphate groups [87]. The N-sulphate groups of heparin are necessary for its heparanase inhibitory activity but can be replaced by an acetyl group as long as the O-sulphate groups are retained and complete desulphation of heparin diminishes all heparanase activity [87].
Similarly, endo sulfatases which include sulf-1 and sulf-2 are also responsible for altering the structure of HS by removal of the 6-O sulphates. This has profound effects on cell signalling pathways which are important for cell function and development [88,89].
The most abundant vascular PGs include decorin and biglycan which are characterized by either one (decorin) or two (biglycan) CS/DS chains [90]. DS is involved in the inhibition of thrombin by HCII [40]. Circulating DS has previously been detected in the plasma of pregnant women at term and in neonatal cord blood at delivery although the levels detectable in the systemic circulation are negligible in comparison with the localised expression of DS within the placenta [91].
Both decorin and biglycan have been shown to be expressed in the endothelial cells of the fetal capillaries and both mRNA and protein expression is reported to be decreased in idiopathic FGR placentae with decorin also significantly reduced in PE-affected pregnancies [67,92,93]. They speculate that because both these PGs are in close proximity to fetal blood, their location allows convenient contact with HCII to inhibit thrombin and thus prevent clot formation. Therefore, the reduction of biglycan and decorin observed in FGR and PE-affected pregnancies could indicate an impairment of this process with less DS readily available to inhibit thrombin [92,93].
There are currently limited options available for the prevention or treatment of PE. However, a reduction in PG abundance and thus their respective GAGs may be a possible mechanism for the disturbances we observe in coagulation, inflammation and angiogenesis in PE-affected pregnancies. This prompts one to question whether replacing this deficiency in GAGs can aid in the prevention of PE.
Heparin is a highly sulphated pharmacological GAG produced by mast cells and is commonly used as an anticoagulant for the treatment of venous thromboembolism [94]. It is structurally similar to heparan sulphate but differing only in the level of sulphation with heparin being more highly sulphated (Figure 2). Heparin exerts its anticoagulant effect by increasing the rate at which factor X and thrombin bind to antithrombin [95].
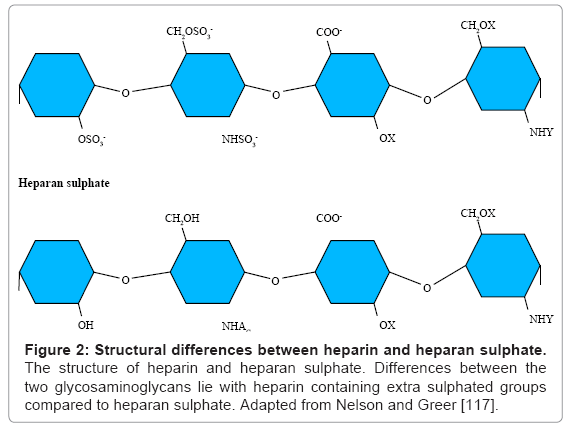
Figure 2: Structural differences between heparin and heparan sulphate. The structure of heparin and heparan sulphate. Differences between the two glycosaminoglycans lie with heparin containing extra sulphated groups compared to heparan sulphate. Adapted from Nelson and Greer [117].
Unfractionated heparin (UFH) has an average molecular weight of 13 to 15 kDa. It does not cross the placenta barrier because of its high molecular weight and therefore does not affect the fetal haemostatic system [96]. However, UFH is associated with significant maternal side effects including osteoporosis and heparin-induced thrombocytopenia [97]. For this reason low molecular weight heparins (LMWH) are thought to be more suitable for use as anticoagulants during pregnancy as they exhibit similar efficacy to UFH with the advantages of an increased half life, improved bioavailability and reduced side effects [98,99].
In fact, due to its small size it is difficult for LMWH to bind simultaneously to AT and thrombin which means that LMWH has reduced ability to inactivate thrombin compared to UFH. In contrast, however, it is this reduced binding capacity that is responsible for the longer half life and lower risk of osteoporosis and heparin induced thrombocytopenia that is observed with LMWH treatment compared to UFH [99]. The biggest problem with the use of heparins during pregnancy, however, relates to the increase risk of bleeding, intrapartum haemorrhage and wound haematoma [100]. Moreover, women receiving antenatal anticoagulants are unable to receive epidural analgesia or spinal anaesthesia due to the risks of epidural or spiral haematoma and the risk of subsequent paralysis [101].
Recent studies have investigated the effects of LMWH in the treatment of pregnancy complications. The Fragmin in pregnant women with a history of Uteroplacental Insufficiency and Thrombophilia (FRUIT) study investigated 139 women with previous hypertensive disease in pregnancy and known thrombophilias. These women were randomised to receive either low dose aspirin or aspirin with the addition of LMWH to determine the rate of recurrence of hypertensive disorders such as PE. The addition of LMWH to aspirin reduced the incidence of recurrent early onset (less than 34 weeks gestation) PE in women with inherited thrombophilia suggesting beneficial effects of using LMWH as a preventative agent (0/70 women on LMWH plus aspirin vs. 6/69 on aspirin alone (risk difference (RD) 8.7%, 95% CI of RD 1.9-15.5, p=0. 012; number needed to treat=12)) [102]. Another randomized control trial investigated the recurrence rate of pregnancy complications such as PE in women without thrombophilia by treating them with LMWH. This study found LMWH treatment to be effective in reducing the recurrence of such pregnancy complications even in women without thrombophilia (3/55 women on LMWH vs. 13/55 controls (OR 0.15, 95% CI 0.03-0.70)) [103]. Together, these two studies suggest that LMWH may exert beneficial effects (in terms of prevention of PE) regardless of the patient’s thrombophilia status.
Most of these studies, however, assume heparins operate via their anticoagulant mechanisms as they assume placental infarction is the predominant pathology. Kingdom et al. undertook a pilot randomized trial investigating the use of UFH in preventing recurrent PE. This study was the first study to investigate the effects of heparin on placental function. Although the sample size was small, it was suggested that UFH was not acting primarily as an anticoagulant as UFH did not prevent placental infarction [104]. In addition, Sobel et al. investigated the non-anticoagulant effects of heparin in normal human and PE placental villi and found both UFH and LMWH heparin promoted in vitro angiogenesis [105]. Importantly, these studies highlight the extensive range of mechanisms in which heparin can exert its effect and thus should redirect pregnancy related heparin research beyond their well established anticoagulant roles.
Although the use of heparins to prevent PE appears promising, the mechanism by which heparin acts within the placenta is largely unknown and is worthy of further investigation in order to maximise the efficacy of pharmacologic GAGs whilst minimising their side effects.
In summary, uncomplicated pregnancies represent a hypercoagulable state with an increase in pro-coagulant activity observed. However, placental thrombosis is rare in these pregnancies which suggest that thrombin generation must be tightly regulated locally within the uteroplacental circulation. This is further validated in PE-affected pregnancies where we observe an exaggerated increase in pro-coagulant activity leading to thrombosis in the uteroplacental circulation. Therefore, the tight haemostatic regulation observed in uncomplicated pregnancies is disturbed in PE.
PGs are abundantly expressed in the placenta and have known anticoagulant, inflammatory and pro-angiogenic properties. Reduced expression of placental PGs, and their associated GAGs, in PE-affected pregnancies may contribute to the increased placental thrombin generation, altered angiogenic and disrupted inflammatory processes seen in this important pregnancy complication. Thus, understanding the mechanism of how altered GAGs may contribute to the pathogenesis of PE is imperative. Investigating and comparing GAG abundance, structure and function between complicated and uncomplicated pregnancies may help to elucidate the complex signalling pathways in which these molecules are involved.
The use of pharmacologic GAGs during pregnancy is becoming increasingly popular with recent randomized controlled trials suggesting that patients treated throughout pregnancy with heparin have a decreased likelihood of developing PE. However, the mechanism by which heparin acts to reduce the likelihood of pregnancy complications is far from certain and deserves further investigation in order to improve the efficacy and minimize the harmful effects of these drugs during pregnancy.