Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2013) Volume 0, Issue 0
GABA is an important neurotransmitter involved in drug abuse and addiction. Accordingly, drugs that elevate brain GABA levels are thought to be promising for the treatment of addiction. In the present study, we report that tiagabine, a selective type 1 GABA transporter inhibitor that has been approved by the U.S. FDA as an anticonvulsant drug, may have therapeutic potential in reducing cocaine use. In vivo brain microdialysis studies demonstrated that systemic administration of tiagabine (3, 10, 20 mg/kg, i.p.) significantly elevated extracellular GABA levels in the nucleus accumbens in a dose-dependent manner. Pretreatment with tiagabine (3, 10 mg/kg, i.p.) significantly inhibited cocaine enhanced electrical brain-stimulation reward. Tiagabine alone, at 20 mg/kg, almost completely inhibited intracranial electrical brain-stimulation behavior. In addition, systemic administration of tiagabine (10, 20 mg/kg) also dose dependently inhibited intravenous cocaine self-administration, but had no effect on cocaine-induced reinstatement of drug-seeking behavior. These data suggest that: 1) elevation of brain GABA levels by tiagabine not only inhibits brain reward function but also attenuates cocaine’s rewarding effects, and 2) tiagabine or other GABA transporter inhibitors may have therapeutic effects in reducing cocaine use, but not in preventing relapse to drug-seeking behavior.
<Keywords: Tiagabine; Cocaine; GABA; Brain-stimulation reward; Self-administration; Reinstatement
Gamma-aminobutyric acid (GABA) is a major inhibitory neurotransmitter in the central nerve system (CNS) which modulates cocaine-taking and cocaine-seeking behavior. It is well documented that cocaine’s rewarding effects are mediated by blocking dopamine transporters (DAT), which causes an increase in extracellular DA level in the nucleus accumbens (NAc) [1]. DA subsequently inhibits NAc medium-spiny (GABAergic) projection neurons and decreases GABA release in the projection areas such as the ventral tegmental area (VTA) and the ventral pallidum (VP) [2,3], two brain regions critically involved in drug reward and addiction [1]. Thus, pharmacological agents that enhance GABAergic neurotransmission in such brain regions should be able to block cocaine’s rewarding effects and thereby reduce cocaine use and abuse [4-9]. Among such drugs, baclofen, a selective GABAB receptor agonist, and γ-vinyl GABA (GVG), an irreversible GABA transaminase inhibitor that elevate extracellular GABA levels, significantly inhibit cocaine self-administration and cocaine-induced reinstatement of drug-seeking behavior [5-8,10]. However, baclofen’s limited therapeutic effects in clinical trials [5] and undesirable toxicities such as visual field defects of GVG [7] prompted us to search for other more effective GABAergic agents for the treatment of cocaine addiction.
Tiagabine is a selective type 1 GABA transporter (GAT1) inhibitor that is widely used as an anti-convulsive drug in humans. The anticonvulsive effects of tiagabine are thought to be mediated by elevated brain extracellular GABA levels [11]. Previous studies with tiagabine for treatment of cocaine addiction have been controversial. In experimental animals, tiagabine was reported to inhibit intravenous cocaine self-administration in baboons at a very low dose (0.1 mg/kg, i.m.) [12] and cocaine-induced reinstatement of drug-seeking behavior in rats at much higher doses (5-10 mg/kg, i.p.) [10]. In contrast to these findings, other studies suggest that tiagabine has no significant effects in either cocaine self-administration in rats [10] or cocaine-induced reinstatement of drug-seeking behavior in baboons [13]. Clinical trials with tiagabine for the treatment of cocaine addiction have also shown different results [14]. Several studies suggest a significant reduction in cocaine intake and/or an increase in cocaine-free urines after tiagabine compared to placebo [15,16], while other studies suggest ineffectiveness of tiagabine in preventing cocaine use in humans [17,18]. The reasons underlying such conflicting findings are unclear. Since significantly different drug doses were used in the above preclinical (0.1-0.3 mg/ kg, i.m. in baboon vs 5-10 mg/kg, i.p. in rats) and clinical studies (4- 24 mg/day, 1-10 weeks), it was hypothesized that optimal doses could be a key issue in evaluation of the therapeutic efficacy of tiagabine for treatment of cocaine addiction. Given that tiagabine is a U.S. FDAapproved anticonvulsant drug while there is no effective medicine approved by the FDA for treatment of cocaine addiction, we further investigated the therapeutic potential of tiagabine for treatment of cocaine addiction in this study. We first used in vivo microdialysis to measure tiagabine-induced changes in brain extracellular GABA levels to find effective drug doses to be tested in the following behavioral experiments. We then observed the effects of tiagabine on basal and cocaine-enhanced electrical brain-stimulation reward, intravenous cocaine self-administration, and cocaine-induced reinstatement (relapse) of drug-seeking behavior in rats. We found that systemic administration of tiagabine, at the doses that elevate brain extracellular GABA levels, significantly inhibits cocaine’s rewarding effects, but has no effect on cocaine-induced reinstatement of drug-seeking behavior in rats, suggesting potential use of tiagabine in reducing cocaine use, but not in preventing relapse to drug-seeking behavior.
Animals
Experimentally naïve male Long-Evans rats (Charles River Laboratories, Raleigh, NC, USA) weighing 250 to 300 g were used for all experiments. They were housed individually in a climate-controlled animal colony room on a reversed light-dark cycle (lights on at 7:00 PM, lights off at 7:00 AM) with free access to food and water. The animals were maintained in a facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International). All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the U.S. National Academy of Sciences, and were approved by the Animal Care and Use Committee of the National Institute on Drug Abuse of the U.S. National Institutes of Health.
Experiment 1: In vivo microdialysis: In vivo microdialysis protocols were as reported previously [20]. Briefly, rats were anesthetized with sodium pentobarbital, and guide cannulae (20 gauge, Plastics One, Roanoke, VA) were surgically implanted into the NAc (AP: +1.7 mm, ML: ± 2.0 mm, DV: -4.0 mm, 6° from vertical), according to the rat brain atlas of Paxinos and Watson (1998). The guide cannulae were fixed to the skull with 4 stainless steel jeweler’s screws (Small Parts Inc., Miami Lakes, FL, USA) and dental acrylic. After 7 days of recovery from surgery, in vivo brain microdialysis was performed. Microdialysis probes were inserted into the NAc 12 hr before the onset of microdialysis to minimize damage-induced neurotransmitter release. Microdialysis samples were collected every 20 min into 10 μl 0.5 M perchloric acid to prevent neurotransmitter degradation. After 1 h of baseline sample collection, 1 of 3 doses of tiagabine (3, 10, 20 mg/kg, i.p.) or vehicle (saline) was administered systemically. After collection, samples were frozen at -80°C.
Quantification of GABA
Concentrations of GABA in the microdialysis samples were determined using HPLC with flourometric detection. The mobile phase consisted of 18% acetylnitrile (v/v), 100 mM Na2HPO4, and 0.1 mM EDTA, pH 6.04. A VeloSep RP-18, 10 cm×3 μm ODS reversed phase column (PerkinElmer Life and Analytical Sciences, Inc., Wellesley, MA) was used to separate the amino acids, and precolumn derivatization of amino acids with o-phthalaldehyde was performed using an ESA Biosciences model 542 autosampler. GABA was detected by an ESA Biosciences Linear Fluor LC 305 fluorescence spectrophotometer. Excitation (Exλ) and emission (Emλ) wavelengths were 336 nm and 420 nm, respectively. The area under the curve (AUC) of the GABA peak was measured using the EZChrom Elite™ chromatography data analysis system (ESA Biosciences, Inc., Chelmsford, MA). GABA values were quantified with an external standard curve (10-1000 nM). The limit of detection for GABA was 0.1-1 pM.
After microdialysis experiments were completed, rats were anesthetized with a high dose of pentobarbital (>100 mg/kg i.p.) and perfused transcardially with 0.9% saline followed by 10% formalin. Brains were removed and placed in 10% formalin for histological verification of microdialysis probe locations in rat brain.
Experiment 2: Intracranial electrical brain-stimulation reward
Surgery: The intracranial electrode implantation surgery and the experimental apparatus were the same as reported previously [20]. Under the same anesthesia as used in Experiment 1, rats were placed in a stereotaxic frame, and a unilateral monopolar stainless-steel stimulating electrode (Plastics One, Roanoke, VA, USA) was placed into the medial forebrain bundle at the anterior-posterior level of the lateral hypothalamus, using standard aseptic surgical and stereotaxic techniques. The implant coordinates for the tips of the electrodes were AP -2.56, ML ± 1.9, and DV -8.6, according to the rat brain stereotaxic atlas of Paxinos and Watson (1998). The electrode was attached to the skull with jeweler’s screws and dental acrylic. A wire leading from the electrode was wrapped around a skull screw to serve as a current return.
General procedure: The general procedures for electrical brain stimulation reward (BSR) were the same as we have reported previously [20]. Briefly, after 7 days of recovery from surgery, rats were allowed to self-train (autoshape) to lever-press for rewarding BSR. Each press on the lever resulted in a 500-msec train of 0.1-msec rectangular cathodal pulses through the electrode in the rat’s medial forebrain bundle, followed by a 500 msec “timeout” in which further presses did not produce brain stimulation.
Rate-frequency BSR procedure: Following establishment of leverpressing for BSR, animals were presented with a series of 16 different pulse frequencies, ranging from 141 to 25 Hz in descending order. At each pulse frequency, animals responded for 2 30-sec time periods (“bins”), after which the pulse frequency was decreased by 0.05 log units. Following each 30-sec bin, the lever retracted for 5 sec. Throughout the experiment, animals were run for 3 sessions a day. The response rate for each frequency was defined as the mean number of lever responses during 2 30-sec bins. Since lever-pressing behavior was variable during the first session (the “warm up” session), but was stable during the second and third sessions, the data from the first session were discarded, and the data from the second and third sessions were designated as the baseline session data and test session data, respectively. The BSR threshold (θ0) was defined as the minimum frequency at which the animal responded for rewarding stimulation. In addition, M50, i.e., stimulation frequency for half maximal reward efficacy, was also used to evaluate the effects of drugs on BSR itself or on cocaine-enhanced BSR. BSR threshold (θ0) and M50 were mathematically derived for each “baseline” run and each “drug” run by analyzing each rate-frequency BSR function generated by a given animal over a given descending series of pulse frequencies using “best-fit” mathematical algorithms. Specifically, each rate-frequency BSR function was mathematically fitted, by iterative computer programs derived from the Gauss- Newton algorithm for nonlinear regression, to 3 different sigmoid curve-fitting mathematical growth models that appear to accurately fit rate-frequency brain-stimulation reward functions [20]. Thus, for each rate-frequency BSR function generated by a given animal over a given descending series of pulse frequencies, 3 solutions for θ0 were obtained. The 3 solutions for θ0 were averaged, to produce a mean θ0 for each rate-frequency BSR function generated by a given animal over a given descending series of pulse frequencies. The mean θ0 values were expressed as means ± S.E.M. Data analyses were performed on percent changes from baseline levels.
Testing the effects of cocaine and/or tiagabine on BSR: Once a baseline θ0 value was achieved (<15% variation over 5 continuous days), the effects of cocaine and/or tiagabine on BSR were assessed. On test days, animals randomly received 1 of 3 or 4 different doses of tiagabine (1, 3, 10, 20 mg/kg i.p.) or vehicle (1 ml 25% 2-hydroxypropyl-β- cyclodextrin) 30 min prior to a cocaine injection (2 mg/kg i.p.). After each test, animals received an additional 5-7 days of BSR re-stabilization until a new baseline θ0 was established. The order of testing for various doses of tiagabine was counterbalanced. The effect of tiagabine on cocaine-enhanced BSR was evaluated by comparing cocaine-induced alterations in θ0 value in the presence or absence of each dose of tiagabine pretreatment.
Experiment 3: Cocaine self-administration and reinstatement test
Cocaine self-administration: Intravenous (i.v.) jugular catheterization and cocaine self-administration experiments were as previously described [9]. After 5-7 days of recovery from jugular catheterization surgery, each rat was placed into a test chamber and allowed to lever-press for i.v. cocaine (1 mg/kg/injection) on a fixedration 1 (FR1) schedule of reinforcement. Each session lasted 3 h. After training for 3-5 days, the subjects were transferred to cocaine self-administration (0.5 mg/kg/injection) under a fixed-ration 2 (FR2) reinforcement schedule until the following criteria for stable cocainemaintained responding were met: less than 10% variability in interresponse interval and less than 10% variability in number of active lever-presses for at least 3 consecutive days. Then each animal randomly received vehicle (saline) or one dose of tiagabine (10, 20 mg/kg, i.p., 30 min prior to test. The effects of tiagabine on cocaine self-administration were evaluated in the same group of rats.
Extinction and testing for reinstatement: After stable cocaine selfadministration was established, the same group of rats was exposed to extinction conditions, during which cocaine was replaced by saline, and the cocaine-associated cue light and tone were turned off. Daily 3 h extinction sessions for each rat continued until that rat lever-pressed less than 10 times per 3 h session for at least 3 consecutive days. After meeting extinction criteria, animals were divided into 3 groups for reinstatement testing. On the reinstatement test day, each rat received either vehicle (saline) or 1 dose of tiagabine (10 or 20 mg/kg, i.p.) 30 min before the reinstatement test. All rats were then given a priming injection of cocaine (10 mg/kg i.p.) immediately before the initiation of reinstatement testing. During reinstatement testing, conditions were identical to those in extinction sessions. Cocaine-induced active lever pressing responses (reinstatement) were recorded, although these responses did not lead to either cocaine infusions or presentation of the conditioned cues. Reinstatement test sessions lasted 3 h.
Statistical Analysis: Data are presented as means ± S.E.Ms. One-way ANOVA for repeated measures was used to determine the significance of the effects of tiagabine on basal and cocaine-enhanced brainstimulation reward, cocaine self-administration and cocaine-induced reinstatement of drug-seeking behavior. Two-way ANOVA for repeated measures over time was used to determine the significance of tiagabineinduced changes in brain extracellular GABA levels. Whenever a significant main effect was found, individual group comparisons were carried out using the Student-Newman Keuls method.
Tiagabine dose-dependently elevates extracellular GABA in the NAc
To determine which doses of tiagabine significantly elevate brain extracellular GABA levels, we used in vivo microdialysis to measure tiagabine-induced changes in GABA in the NAc. Figure 1 shows that systemic administration of tiagabine (3, 10, 20 mg/kg, i.p.) significantly elevated extracellular GABA levels in a dose-dependent manner. Two-way ANOVA for repeated measures revealed a statistically significant treatment main effect (F3,19=9.43, P<0.001), time main effect (F17,323=9.84, P<0.001) and treatment×time interaction (F51,323=5.11, P<0.001). Individual group comparisons revealed a significant increase in GABA after 10 mg/kg (q=4.43, P<0.05) or 20 mg/kg (q=6.61, P<0.01), but not 3 mg/kg (q=0.19, P=NS), tiagabine when compared to vehicle. To further determine whether 3 mg/kg tiagabine-induced short-time increase in GABA was statistically significant, we used oneway ANOVA for repeated measures for the data 1 hr before and 1 hr after 3 mg/kg tiagabine and found that 3 mg/kg tiagabine produced a statistically significant increase in extracellular GABA level (F5,20=5.64, P<0.01). Post-hoc individual group comparisons revealed a significant increase in extracellular GABA at 40 min (q=6.33, P<0.11) after 3 mg/kg tiagabine when compared to baseline before tiagabine administration.
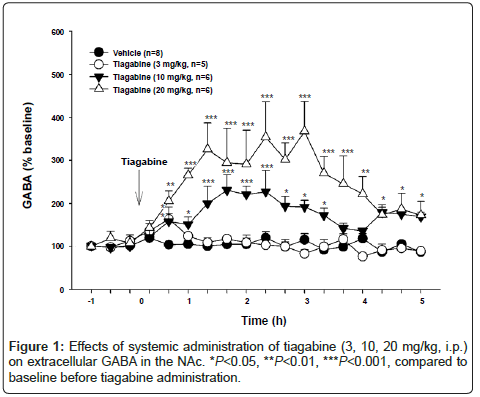
Figure 1: Effects of systemic administration of tiagabine (3, 10, 20 mg/kg, i.p.) on extracellular GABA in the NAc. *P<0.05, **P<0.01, ***P<0.001, compared to baseline before tiagabine administration.
Tiagabine attenuates cocaine-enhanced brain-stimulation reward (BSR)
Figure 2A shows representative rate-frequency function curves for BSR, indicating BSR stimulation threshold (θ0 Hz), M50 and Ymax (maximal lever presses/30 sec). Figure 2B shows representative BSR rate-frequency function curves, illustrating the effects of cocaine and tiagabine on BSR function. Cocaine, at 2 mg/kg, produced a significant enhancement of BSR, as indicated by the leftward shift in the rate-frequency function curve, reflecting lowered BSR threshold (θ0) values. This cocaine-enhanced BSR was attenuated by tiagabine (3 mg/kg, i.p., 30 min prior to cocaine administration). Figure 2C shows the averaged effects of cocaine and tiagabine on BSR threshold. Oneway ANOVA for repeated measures revealed a statistically significant tiagabine treatment main effect (F3,30=5.20, p<0.01). Individual group comparisons revealed that 2 mg/kg cocaine significantly enhanced electrical BSR (vehicle versus 2 mg/kg cocaine: q=9.41, p<0.001), an effect that was significantly attenuated by 3 mg/kg (q=4.75, p<0.01) or 10 mg/kg (q=3.86, p<0.01), but not by 1 mg/kg (q=0.177, p=NS) tiagabine. Figure 2D shows the effects of tiagabine alone, illustrating that tiagabine (1, 3, 10 mg/kg) did not significantly alter electrical BSR by itself (F3,21=1.92, P=NS). However, at 20 mg/kg, tiagabine completely inhibited electrical brain-stimulation behavior in the absence or presence of cocaine (data not shown because no θ0 values were given in such experimental conditions). Figures 2E and 2F show the effects of tiagabine on Ymax, illustrating that tiagabine dose-dependently lowered Ymax (Figure 2E: F3,30=3.76, P<0.05; Figure 2F: F3,21=3.71, P<0.05). Post-hoc individual group comparisons revealed a significant reduction in Ymax after 10 mg/kg (Figure 2E: q=4.63, P<0.05; Figure 2F: q=4.43, P<0.05), but not 1 or 3 mg/kg tiagabine.
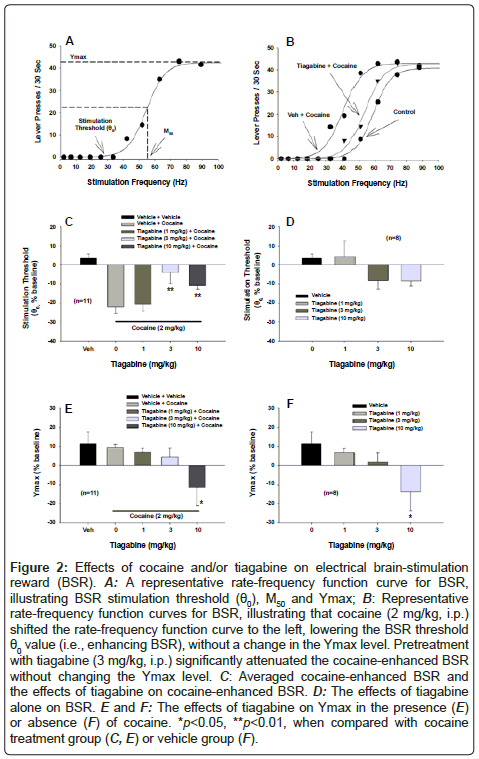
Figure 2: Effects of cocaine and/or tiagabine on electrical brain-stimulation reward (BSR). A: A representative rate-frequency function curve for BSR, illustrating BSR stimulation threshold (θ0), M50 and Ymax; B: Representative rate-frequency function curves for BSR, illustrating that cocaine (2 mg/kg, i.p.) shifted the rate-frequency function curve to the left, lowering the BSR threshold θ0 value (i.e., enhancing BSR), without a change in the Ymax level. Pretreatment with tiagabine (3 mg/kg, i.p.) significantly attenuated the cocaine-enhanced BSR without changing the Ymax level. C: Averaged cocaine-enhanced BSR and the effects of tiagabine on cocaine-enhanced BSR. D: The effects of tiagabine alone on BSR. E and F: The effects of tiagabine on Ymax in the presence (E) or absence (F) of cocaine. *p<0.05, **p<0.01, when compared with cocaine treatment group (C, E) or vehicle group (F).
Figure 3 shows that systemic administration of tiagabine (10-20 mg/ kg) significantly inhibited cocaine self-administration in rats. Figure 3A shows representative records of cocaine self-administration (i.e., infusions) after vehicle or tiagabine in the same rat tested at different days, illustrating that 20 mg/kg tiagabine significantly inhibited cocaine self-administration, an effect that lasted for about 1 hour. Figure 3B shows the averaged numbers of cocaine infusions and inactive lever responses in 3 hr session after different doses of tiagabine, illustrating that tiagabine dose-dependently inhibited cocaine self-administration (one-way ANOVA, treatment main effect, F2,18=5.03, P<0.05). Figure 3C shows that tiagabine also significantly decreased active lever presses (treatment main effect, F2,18=6.90, P<0.05), but had no effect on inactive lever presses (F2,18=0.790, P=NS).

Figure 3: Effects of tiagabine on cocaine self-administration. A: Representative cocaine self-administration records after vehicle or 20 mg/kg tiagabine treatment in the same rat. B: Total numbers of cocaine infusions after tiagabine pretreatment. C: Active and inactive lever responses after tiagabine pretreatment. * P<0.05, compared with vehicle control group.
Figure 4 shows the effects of tiagabine (10-20 mg/kg) on cocaine induced reinstatement of drug-seeking behavior. Figure 4A shows representative cocaine-seeking behavior (i.e., active lever presses) in two different rats, illustrating that tiagabine had no effect on cocaineseeking behavior induced by cocaine priming (10 mg/kg). Figures 4B and 4C show the averaged active and inactive lever responses on the last day of cocaine self-administration (SA), the last day of extinction (Ext), and the following reinstatement test day, illustrating that tiagabine did not significantly alter cocaine-induced reinstatement of drugseeking behavior (Figure 4B: one-way ANOVA, treatment main effect, F2,18=0.08, P=NS; Figure 4C: F2,18=0.54, P>0.05).
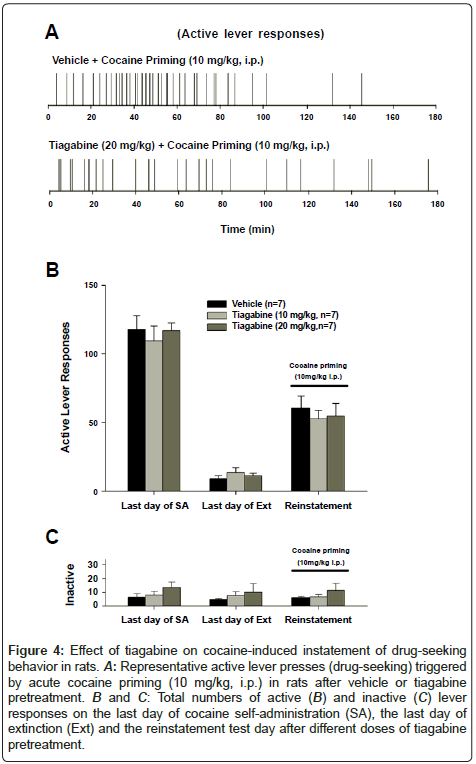
Figure 4: Effect of drug-seeking on cocaine-induced instatement of drug-seeking behavior in rats. A: Representative active lever presses (drug-seeking) triggered by acute cocaine priming (10 mg/kg, i.p.) in rats after vehicle or tiagabine pretreatment. B and C: Total numbers of active (B) and inactive (C) lever responses on the last day of cocaine self-administration (SA), the last day of extinction (Ext) and the reinstatement test day after different doses of tiagabine pretreatment.
The present study demonstrates that systemic administration of tiagabine (3, 10, 20 mg/kg) significantly elevates extracellular GABA levels in the NAc in a dose-dependent manner. The dose of 3 mg/kg tiagabine appears to be the minimal effective dose to elevate brain extracellular GABA level, while 10-20 mg/kg tiagabine produced a robust (200-400%) and long-term (4-5 hrs) increase in extracellular GABA levels. Based on this finding, we used the same doses of tiagabine in the following behavioral experiments.
Electrical brain-stimulation reward (BSR) is a reliable and sensitive animal model for assessing brain reward function and the reward effects of addictive drugs [21,22]. Here, we found that cocaine significantly decreased BSR thresholds, indicative of summation between the reward induced by the electrical brain stimulation and that produced by cocaine. Strikingly, this cocaine-enhanced BSR was significantly attenuated by tiagabine (3, 10 mg/kg) pretreatment. Further, tiagabine itself, at 20 mg/kg but not lower doses, produced cessation of BSR behavior in the majority of rats tested. These findings suggest that elevation of brain extracellular GABA levels not only attenuated cocaine’s rewarding effects, but also inhibit brain reward function by itself. This is consistent with a previous report that GVG, a GABA transaminase inhibitor, also significantly inhibits BSR and cocaine-enhanced BSR by a similar mechanism – elevation of extracellular GABA levels in the brain [23].
Congruent with tiagabine’s antagonism of cocaine-enhanced BSR, tiagabine also dose-dependently inhibited cocaine self-administration under FR2 reinforcement. Since intravenous self-administration is a commonly used animal model to measure drug’s rewarding effects [24-26], the present finding, combined with the finding in the above BRS experiment, suggests that pretreatment with tiagabine attenuates cocaine’s rewarding effects. Strikingly, the effective dose (20 mg/ kg) is much higher than those tested previously in cocaine selfadministration [10,12]. This finding is consistent with a previous report that 10 mg/kg tiagabine marginally inhibited cocaine-taking behavior [10], but is different from another reports that 0.1 mg/kg tiagabine significantly inhibited cocaine self-administration in baboon [12]. The mechanisms underlying these different findings are unclear. It could be related to different experimental conditions or species. Although 3 mg/kg tiagabine appears to be the minimal effective dose in elevating brain extracellular GABA levels in the present study, we cannot exclude such a possibility that lower doses of tiagabine produces a significant increase in extracellular GABA in other brain regions in baboon. We also note that the effective tiagabine dose (20 mg/kg) observed in cocaine self-administration is significantly higher than those used in the above BSR experiment (3, 10 mg/kg). This could be related to the fact that the accumulative cocaine dose (0.5 mg/kg/injection×50 infusions=25 mg/kg) in the present self-administration experiment was much higher than those used in the above BRS experiment (2 mg/kg). Thus, higher doses of cocaine intake may counteract the therapeutic effects of tiagabine.
Although tiagabine alone, at 10-20 mg/kg, significantly lowered Ymax values in the above BSR experiment, suggesting locomotor inhibition or sedative effects, we do not think such sedation underlying the reduction in the above cocaine self-administration since the same doses of tiagabine altered neither inactive lever response in cocaine self-administration experiment (Figure 3B) nor active and inactive lever responses in the reinstatement experiment (Figure 4B). A possible interpretation is that cocaine-induced locomotion-stimulating effects may functionally counteract tiagabine-induced locomotor inhibition at high doses.
Growing evidence suggests that a DA-GABA mechanism in the NAc may underlie cocaine’s rewarding effects [8,27]. Anatomically, the majority of neurons in the NAc are medium-spiny GABAergic output neurons, which receive DA projections from the VTA and project to the VTA and the ventral pallidum [28,29]. Cocaine elevates extracellular DA by blocking DA transporters. It is well documented that DA produces a net inhibitory effect on striatal medium-spiny GABAergic neurons [30,31], predominantly by activation of D2-like DA receptors [32], which causes a reduction in GABA release in the projection areas such as the VTA and ventral pallidum [3,33]. Such a reduction in GABA release in the VTA may further increase VTA DA neuron activity by a disinhibition mechanism [2]. Therefore, decreased NAc-VTA and/or NAc-VP GABA transmission may constitute a final common pathway underlying drug (including cocaine) reward. Based on this, elevation of extracellular GABA levels by tiagabine, particularly in the VTA and VP, would functionally antagonize cocaine’s action on VTA DA neuron activity and VP GABA release; thereby attenuating cocaine’s rewarding effects, as assessed by intracranial BSR and intravenous cocaine selfadministration in the present study. We note that tiagabine itself produced a significant and dose-dependent increase in extracellular GABA levels in the NAc, which, in theory, should enhances, rather than attenuates, cocaine-induced reduction in VTA/VP GABA release. A possible interpretation is that the reduction in cocaine and brain reward function as observed in the present study could be a final net effect of multiple actions in different brain regions after tiagabine administration. Clearly, more studies are required to address this issue.
An unexpected finding is that that tiagabine, at 10-20 mg/kg, failed to alter cocaine-induced reinstatement of drug-seeking behavior. This is consistent with a previous report with tiagabine (0.3 mg/kg, i.m.) in baboons [24], but conflict with another report demonstrating that 5-10 mg/kg tiagabine significantly inhibits cocaine- or foodinduced reinstatement of reward-seeking behavior in rats [10]. The reasons underlying such conflicting findings are unclear. Whatever the reasons, the present finding is consistent with a current view that cocaine-induced reinstatement of drug-seeking behavior is mediated predominantly by a NAc glutamate, but not DA or GABA, mechanism [34,35]. That is, chronic cocaine produces an enduring reduction in basal glutamate release in the NAc during cocaine withdrawal or abstinence, while cocaine priming stimulates presynaptic glutamate release from neuronal terminals in the VAT and the NAc. These findings suggest that both the reduced basal and enhanced glutamate release may constitute a neurobiological substrate of relapse to drug-seeking behavior [8,34,35]. We have previously reported that elevation of brain GABA levels may contribute to reduced basal glutamate release in the NAc [36]. However, pharmacological elevation of extracellular NAc GABA by GVG failed to block cocaine-enhanced NAc DA [37,38], suggesting that tiagabine may also fail to alter cocaine-induced increase in synaptic glutamate release and subsequent reinstatement of drug-seeking behavior. More studies are required to test this hypothesis.
In conclusion, the present study shows that systemic administration of tiagabine significantly and dose-dependently elevates extracellular GABA levels in the NAc, inhibits cocaine-enhanced brain-stimulation reward and intravenous cocaine self-administration, but has no effect on cocaine-induced reinstatement of cocaine-seeking behavior in rats. The conflicting findings observed in the previous studies in both experimental animals and human addicts may be importantly related to the drug doses used in those studies. The present findings suggest that tiagabine may have therapeutic potential in attenuating cocaine’s rewarding effects, thereby decreasing cocaine use and abuse, but might have limited effect in preventing relapse to drug-seeking behavior.
This research was supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health, Department of Health and Human Services.