Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 2, Issue 3
Human opiorphin inhibits enkephalin-inactivating ectopeptidases to produce analgesic and antidepressant-like effects in standard murine models via activation of μ and/or δ opioid pathways. It is an endogenous peptide regulator of enkephalin bioavailability. Opiorphin molecule, a QRFSR-peptide, is thus a promising prototype for the design of an improved class of analgesics. The major limitation on the clinical use of peptide drugs is their rapid degradation by circulating peptidases. Our goal was, therefore, to search for functional derivatives of opiorphin with improved metabolic stability. In order to identify the functional amino acid groups required for opiorphin inhibitory potency toward both AP-N and NEP human ectopeptidases, we used the Structure-Activity Relationship (SAR) method. From this data, a series of opiorphin derivatives was designed and selected. The best performing compound then underwent a complete metabolic profile using in vitro kinetic models. Finally, its safety profile relative to the native peptide as well as its efficacy in an in vivo rat model was evaluated. We demonstrated a tight structural selectivity in the functional interaction of opiorphin with both human NEP and AP-N targets by SAR studies. Nevertheless, we found that the addition of an N-terminal Zn-chelating group, a Cys-thiol group and the replacement of the first labile peptide bond by a polyethylene surrogate, a [CH2]6 linker,and, finally, the substitution of Ser4 by Ser-O-[CH2]8, results in a high performing C-[(CH2)6]-QRF[S-O-[CH2]8]-R peptidomimetic product. This designed opiorphin analog shows reinforced inhibitory potency toward human AP-N activity (more than 10-fold increase) and NEP activities (more than 40-fold increase) relative to the QRFSR native peptide. It also has increased metabolic stability in human plasma and yet retains full analgesic activity in the behavioral formalin-induced rat pain model. C-[(CH2)6]-QRF[S-O-[CH2]8]-R thus represents a very attractive and promising analgesic drug-candidate.
<Keywords: Opiorphin; Inhibitor of enkephalin-inactivating peptidases; Functional peptidomimetics; Analgesic effect; Human opioid pathway
Opiorphin QRFSR-peptide is an endogenous human regulator that was discovered using a functional biochemical approach [1,2]. Its characterization demonstrated that it is an authentic physiological dual inhibitor of Zn-dependent metallo-ectopeptidases, neutral endopeptidase (NEP EC3.4.21.11) and aminopeptidase N (AP-N EC3.4.11.2). These enzymes are implicated in the rapid inactivation of endogenous circulating opioid agonists, namely the enkephalins. As a consequence, opiorphin improves the specific binding and affinity of enkephalin-related peptides to membrane opioid receptors [3]. The enkephalin neuropeptides play key roles in the control of nociceptive transmission and in the modulation of the activity of cerebral structures governing the motivation and the adaptive balance of emotional states [4-8]. By increasing the half-life of circulating enkephalins, opiorphin, at systemically or centrally active doses (1-2 mg/kg I.V. or 5-10 μg/kg I.C.V.), produces analgesia in various murine models of pain [1,9,10]. At equivalent doses, opiorphin also exerts antidepressant-like effects in the standard model of depression, the forced swim test [11,12]. All opiorphin-induced effects are specifically mediated via endogenous enkephalin-related activation of μ and/or δ opioid pathways.
The discovery of opiorphin is the first demonstration of the existence of a physiological regulator of enkephalin bioavailability in humans. As an upstream modulator of opioid pathways in humans, it is thus of major interest from a therapeutic points of view. Indeed, endogenous human opiorphin appears to intervene in the process of adaptation mediated by enkephalins that is associated with nociception. As a consequence, opiorphin is a promising template for the design of a new class of drug-candidates able to efficiently alleviate a number of severe and chronic pain syndromes, without morphine side effects. The actions of opiorphin could be induced at a specific opioid receptor restricted pathway dynamically stimulated by natural effectors, such as enkephalins, that are recruited according to the nature, duration and intensity of the stimulus. This mechanism of action avoids excessive stimulation of ubiquitously distributed opioid receptors and prevents serious side effects such as respiratory depression, sedation, constipation, physical and psychic dependence and tolerance that have been reported in the case of μ-opioid agonists. We previously demonstrated that opiorphin subchronic intake does not develop significant abuse liability or antinociceptive drug tolerance. In addition, anti-peristalsis is not observed [10].
A major limitation to the clinical use of peptide drugs, however, is their rapid degradation by circulating peptidases and the limited permeation of peptides across biological barriers. In order to search for functional derivatives of opiorphin endowed with improved half-life stability and bioavailability properties a Structure-Activity Relationship (SAR) study on opiorphin was first carried out. Then opiorphin analogs were tested for their inhibitory potency against the two membrane-anchored human ectoenzymes, NEP that has both endopeptidase and carboxydipeptidase activities and AP-N, by using selective fluorescence-based enzyme in vitro assays [13-15]. Comparative degradation kinetics were done using experimental in vitro systems to evaluate metabolic half-life in human plasma, which is a reliable prediction model for in vivo stability. Metabolic stability parameters in human liver microsomes were also determined.
The final aim of the research described here was to design and analyze functional analogs of opiorphin that display in vivo bioavailability properties superior to the native peptide, in particular, an increase in circulating peptidase resistance and in permeation across epithelial and endo-epithelial membrane barriers, without affecting in vitro and in vivo biological properties, namely, selective inhibition of human NEP and AP-N ectoenkephalinases and potent inhibition of pain behavioral responses in rat model.
Chemicals
All peptides, human opiorphin and opiorphin derivatives, were synthesized by Genosphere Biotechnologies (Paris-France). Analytical RP-HPLC and electrospray MS confirmed the purity (≥ 95%) and molecular mass of the synthesized peptides.
FRET-based Enzyme in vitro assays
Formal kinetic analysis was performed for each assay using realtime fluorescence monitoring of specific substrate hydrolysis.
Sources of the human ectopeptidases: Human recombinant NEP and human recombinant AP-N (devoid of their respective N-terminal cytosol and transmembrane segment) were purchased from R&D Systems (France) and used as a pure source of peptidases. Membraneanchored NEP and AP-N expressed by a human cell line in culture (serum-free medium), namely, LNCaP epithelial prostate cells, were also used as a source of native ectopeptidases. Human recombinant DPPIV (dipeptidyl amino peptidase IV) and human recombinant ECE- 1 (endothelin converting enzyme), purchased from R&D Systems, were used to assess compound specificity.
Substrates and inhibitors: in vitro, CarboxyDi- and Endo- Peptidase NEP activities were assayedby measuring the breakdown of the following synthetic selective substrates: i, Abz-dR-G-L-EDDnp FRETpeptide, an internally quenched fluorescent substrate specific for NEPEndoPeptidase activity, was synthesized by Thermo-Fisher Scientific (Germany); ii, Abz-R-G-F-K-DnpOH FRET-peptide, an internally quenched fluorescent substrate specific for NEP-CarboxyDiPeptidase activity, was synthesized by Thermo-Fisher Scientific (Germany) [13]; iii, Mca-R-P-P-G-F-S-A-F-K-(Dnp)-OH FRET-peptide (Mca-BK2), an intramolecularly quenched fluorogenic peptide structurally related to bradykinin, which is a selective substrate for measuring NEP and ECE activity, was purchased from R&D Systems; iv, G-P-7amido-4- Mca (G-P-Mca), a selective substrate for measuring DPPIV activity, was purchased from Sigma-Aldrich (France).
FRET is the distance-dependent transfer of energy from a donor fluorophore (Abz=ortho-aminobenzoyl or Mca=7-methoxycoumarin 4-yl-acetyl) to an acceptor fluorophore (DnpOH=2,4-dinitrophenyl or EDDnp=2,4-dinitrophenyl ethylenediamine).
The modified tritiated substance P ([(3,4,3H)Pro2-Sar9-Met(O2)11]- SP, NEN-PerkinElmer) was also used to assess for the specific endopeptidase activity of membrane-bound human NEP under relevant biological conditions of measurement [1,16].
H-alanine-AMC (AMC=amino-methyl-coumarin), Ala-AMC, a fluorogenic substrate for measuring aminopeptidase activity was purchased from Bachem (Switzerland).
Measurement of Ectopeptidase Activities using 96-well fluorimetric assays: Under conditions of initial velocity measurement (steady state), hydrolysis of substrates was measured by real-time monitoring of their metabolism rate by the respective recombinant and membrane-bound peptidases, in the presence and absence of tested inhibitory compound (concentrations ranging from 0.01 to 100 μM).
Measurement of NEP-endopeptidase activity using FRET specific peptide-substrate, Abz-dR-G-L-EDDnp: Using the black half-area 96 well micro-plate, the standard reaction consisted of enzyme (12 ng) in 100 mM Tris-HCl pH 7 containing 200 mM NaCl and 0.05% Brij 35 (100 μl final volume). The substrate (15 μM final concentration) was added after preincubation for 10 min at 28°C and the kinetics of appearance of the fluorescent signal (RFU) was directly analyzed for 20-40 min at 28°C (2 to 3 min interval successive measures) by using a fluorimeter micro-plate reader (monochromator Infinite 200-Tecan) at 320 nm and 420 nm excitation and emission wavelengths, respectively.
Measurement of NEP-Carboxy DiPept idase activity using FRET specific peptide-substrate Abz-R-G-F-K-DnpOH: Using the black halfarea 96 well microplate, the standard reaction consisted of enzyme (2.5 ng) in 100 mM Tris-HCl pH 6.5 containing 50 mM NaCl and 0.05% Brij 35 (100 μl final volume). The substrate (4 μM final concentration) was added after pre incubation for 10 min and the kinetics of appearance of the fluorescent signal (RFU) was directly analyzed for 20-40 min at 28°C (2 to 3 min-interval successive measures) using the fluorimeter reader at 320 nm excitation and 420 nm emission wavelengths.
In addition, the intra-molecularly quenched fluorogenic peptide, Mca-BK2 (2.5 μM final concentration), was submitted to hydrolysis by 2 ng rhNEP under the same experimental conditions as those described above. Under these conditions the hNEP-enzyme acted upon Mca-RP- P-G-F-S-A-F-K-(Dnp)-OH as a CarboxyDiPeptidase preferentially cleaving the A-F bond but also as an EndoPeptidase cleaving the G-F bond.
To measure ECE1-ectopeptidase activity, the same protocol described previously was applied except that Mca-BK2 substrate was used at 7.5 μM final concentration and rhECE1 at 5 ng final concentration.
Measurement of DPPIV activity using FRET specific peptidesubstrate G-P-7amido-4-Mca: Using the black half-area 96 well microplate, the standard reaction consisted of enzyme (7 ng) in 100 mM Tris-HCl pH 8 (100 μl final volume). The substrate (5 μM final concentration) was added after preincubation for 10 min and the kinetics of appearance of the fluorescent signal (RFU) was directly analyzed for 20-40 min at 28°C (2.3 min-interval successive measures) using the fluorimeter reader at 380 nm excitation and 460 nm emission wavelengths.
Measurement of AP-N-ectopeptidase activity using Ala-AMC substrate: Using the black half-area 96 well microplate the standard reaction consisted of enzyme (3.5 ng) in 100mM Tris-HCl pH 7.0 (100 μl final volume). The Ala-AMC substrate (20 μM final concentrations) was added after preincubation for 10 min at 28°C and the kinetics of appearance of the signal was monitored for 20-40 min at 28°C using the fluorimeter reader at 380 nm excitation and 460 nm emission wavelengths.
Measurement of membrane human NEP-endopeptidase activity using tritiated substance P substrate: the method used was previously described in Wisner et al. and Rougeot et al. [1,16].
The background rate of substrate autolysis, representing the fluorescent signal obtained in the absence of enzyme, was subtracted to calculate the initial velocities in RFU (Relative Fluorescent Unit)/ min. Data were analyzed using Magellan 6.0 software to evaluate initial velocities and with Excel Microsoft software. IC50 estimates were obtained from a sigmoidal curve fit to a plot of % inhibitory activity versus log inhibitor concentration, using Prism software. For each curve, inhibitors were tested across a range of concentrations differing in half log unit increments.
in vitropharmacokinetic and metabolic studies: Human blood was collected in pre-chilled tubes containing 1% sodium citrate (buffered at pH 7) and kept at 4°C. The plasma was collected after centrifugation at 400 × g for 30 min at 4°C, then aliquoted and stored at -80°C.
Pharmacokinetic (PK) experiments: After thawing, plasma were again centrifuged for 30 min at 4000 rpm and +4°C, filtered (0.45 μm), and distributed at 4°C in Minisorb tubes (Nunc, Dutscher, France) at 500 μl aliquots for each kinetic point.
Peptide solutions were extemporaneously prepared in order to add the appropriate concentration of peptidein a volume of 10 μl, thus avoiding dilution of the plasma. The plasma peptide solutions were then mixed and incubated in a shaking water bath at 37°C with a continuous and slight shaking for the preset kinetic time period. The reaction was stopped by cooling the tubes simultaneously in ice and by the addition of 0.1N final concentration of HCl. For opiorphin PK experiments, a mixture of 1 μg or 40 μg QRFSR-peptide, containing 100 or 500.103 cpm QR [3H-F] SR (3.6 Ci/mmole, CEA-Saclay), was used. Controls, in which protease free human plasma (Methanol/TFA extract) is substituted for fresh plasma, were included. For certain experiments, different inhibitors of plasma peptidases were added immediately prior to the addition of opiorphin-peptide, bestatin, an inhibitor of aminopeptidases or GEMSA and an inhibitor of carboxypeptidase B.
All samples were stored at -80°C until subjected to Sep-Pak extraction and RP-HPLC chromatography.
C18 Solid-phase extraction: Acidified (HCl 0.1N final concentration) and clarified biological samples were applied to C18-SepPak cartridges (Waters, France) preconditioned with three successive cycles of methanol (Lichrosolv, Merck) and pure water and ultimately maintained in 0.1% TFA-water. After applying the samples to the top of the cartridge and washing with 0.1%TFA-water (5 ml), the analytes were eluted with 100% methanol containing 0.1% TFA (5 ml). The fractions were collected at 4°C, frozen at -80°C and then lyophilized at -110°C for 48 h. Under these conditions, recovery of the marker QR [3H-F] SR-opiorphin, added to plasma samples, was 76 ± 5 % (mean ± SD for n=20).
Finally, dried extracts were re-suspended in 250 μl pyrolyzed water at 4°C then centrifuged 30 min at 4000 rpm and +4°C to quantify opiorphin-related components by radioactivity measurement (radiometer Wallac, PerkinElmer) and RP-HPLC in conjunction with PDA and radiometer analyses and/or ELISA-Opiorphin immunoassays.
Reverse phase C18-HPLC Chromatography: RP-HPLC, coupled with online PDA (224 nm) and radiometric (150-TR PerkinElmer) detection, was used to separate, identify and semi-quantify the different opiorphin-related molecular forms contained in human plasma extracts from in vitro PK experiments. Reversed Phase-High Performance Liquid Chromatography (RP-HPLC) used a C18-bonded stationary phase and an acetonitrile mobile phase in the presence of 0.1% trifluoroacetic acid (TFA, Sigma-France).
The re-suspended extracts (equivalent to 100 μl initial plasma volume), obtained during the above-described procedures, and were applied to the top of the C18/RP-HPLC analytical column (150×4.5 mm Luna 5 μ Phenomenex-France) under TFA 0.1%-water solvent equilibrium conditions. The various components were eluted and isolated according to their hydrophobic characteristics, in a 25- min linear gradient from 0% to 50% acetonitrile (Lichrosol, Merck), containing 0.1% TFA at a 1 ml/min flow rate (Surveyor HPLC system, Thermo Scientific-France). The entire HPLC system was thermoregulated at 12°C. Each fraction (1 ml) was collected and lyophilized at -110°C for 48 h. Each chromatographic profile was driven, integrated and analyzed by the ChromQuest software. The peak height values of each peak of interest as well as those for a defined inner standard peak were calculated. Eluted fractions were collected at a 1 min timeinterval. Each fraction was lyophilized at -110°C for 48 h. In opiorphin PK experiments, the content of radioactivity of each sample, i.e., crude plasma, plasma extracts, HPLC fractions, was determined to evaluate the recovery of each processing step.
The opiorphin-like content of samples (SepPak extracts and/or HPLC fractions) was also measured using a quantitative and specific immunoassay (competitive-ELISA) developed in the laboratory [17].
Immunoassay for opiorphin: The recently published protocol was used to assess the opiorphin-like content of samples [17]. Optimized assay conditions are summarized as follows: For the coating, 40 ng of the Y-[(CH2)12]-QRFSR peptide per 200 μl coating buffer (100 mM potassium phosphate, PH 7.1) were added to individual wells on a 96- well micro-titration plate and incubated overnight at +4°C with light shaking. In parallel, 100 μl of standard or samples, that were serially diluted 2-fold with incubation buffer (200 mM Tris-HCl, pH 7.5+150 mM NaCl+0.1% Tween 20+0.1% bovine serum albumin), were preincubated in Screen Mates tubes (Matrix, Thermo Scientific-France) overnight at 10°C, in the presence of 100 μl anti-opiorphin antibody diluted at 1/80 000. The following day, after washing 5 times with washing buffer (1 tablet PBS-Sigma in 200 ml pure pyrolyzed water + 0.1% tween 20), 250 μl of saturation buffer (20 mM Tris-HCl, pH 7.5+150 mM NaCl+0.1% Tween 20+0.5% gelatin) were added to the individual coated-wells and incubated for at least 1 h at 20°C. Then, after washing, 100 μl of the pre-incubated immunological reaction were transferred onto the coated and saturated micro-titration plates and incubated 1.30 h at 10°C in a humid atmosphere. After washing, 100 μl of the anti-rabbit IgG conjugated to HRP (Pierce, ThermoScientific-France), diluted in Tris buffer (20 mM Tris-HCl, pH 7.5+150 mM NaCl+0.1% Tween 20+0.1% BSA) at 1/3 000, were added to each well and incubated for 1 h at 20°C. After incubation an ultimate wash was performed and 100 μl of the HRP chromogenic substrate (StepUltraTMB-ELISA, ThermoScientific-France) were added and incubated for 30-45 min at 20°C. Finally, the reaction was stopped by adding 100 μl 4N H2SO4. Plates were read at 450 mm with a microplate spectrophotometer (Infinite M200, Tecan-France) and the results were successively analyzed with Magellan (Tecan), Prism GraphPad (La Jolia, USA) and Excel Microsoft softwares.
in vivo studies using a rat pain model
Animals: Male Wistar rats (Harlan, France) weighing 250-280 g were used in this study. After a 7-day acclimatization period, they were weighed and randomly housed according to the treatment groups in a room with a 12 h alternating light/dark cycle (9:00 pm/9:00 am) and controlled temperature (21 ± 1°C) and hygrometry (50 ± 5%). Food and water were available ad libidum. They were experimentally only tested once.
Behavioral tests, care and euthanasia of study animals were in accordance with guidelines of the European Communities Directive 86/609/EEC and the ASAB Ethical Committee for the use of laboratory animals in behavioral research (Animal Behaviour, 2006; 71:245-53). The study protocol was approved by the local ethics committee (Comité d’Ethique Lorrain en Matière d’Expérimentation Animale) with the agreement no. CELMEA-2012-0021 obtained on December 6, 2012.
Chemicals: Opiorphin analog (Genosphere Biotechnologies, France) was dissolved in vehicle solution (55% of PBS 100 mM-45% of Acetic acid 0.01N) and systemically (I.V.) injected, 10 to 15 min prior to the behavioral tests, at doses ranging from 0.5 to 2 mg/kg body weight. Morphine HCl (Francopia, France) was dissolved in saline (0.9% sodium chloride in distilled water) and injected I.V. 15 min before the behavioral test, at 2 mg/kg dose. All drugs were administered in a volume of 1 ml/kg body weight.
The Formalin Test: The previously prescribed protocol [1,10,16] was used to assess the analgesic potency of opiorphin analog in a chemical-induced inflammatory pain model. Groups of 8 rats were used for each experiment. 50 μl of a 2.5% formalin solution was injected under the surface of the left hind paw 10-15 min after I.V. injection of opiorphin analogs, morphine or vehicle. The duration of formalin-injected paw licking and the number of inflamed paw flinches and body tremors were recorded for a period of 60 min after formalin administration. The behavioral scores were expressed as means ± standard error of the mean (SEM) for n=8 rats.
Statistical Evaluation: The significance of differences between groups was evaluated using the Kruskal-Wallis one-way analysis of variance (KWT, a non-parametric method) for comparison between several independent variables across the experimental conditions. When a significant difference among the treatments was obtained, the Mann-Whitney post hoc test (MWT) was applied to compare each treated group to the control one. For all statistical evaluations, the level of significance was set at P < 0.05. All statistical analyses were carried out using the software StatView®5 statistical package (SAS, Institute, Inc., USA).
Structure-activity relationship study
In order to identify the amino acid residues or functional groups required for opiorphin inhibitory potency toward both AP-N and NEP human ectopeptidases, the molecular relationship of structure to activity, namely Structure-Activity Relationship (SAR), of opiorphin native peptide was first investigated. The inhibiting activity of each modified compound was evaluated toward human recombinant NEP (rh-NEP) and AP-N (rh-AP-N), the residual enzyme activity was measured by continuous fluorimetric assays in the presence of specific fluorescent substrate.
Our findings, associated with a previously reported Ala substitution scanning study [18] show:
The importance of the N-terminal amine group of the NH2-QRFSR peptide in the inhibitory potency of opiorphin toward rhAP-N. Indeed, the acetylation (Ac-QRFSR) or pyroglutamylation (pGlu1-RFSR), the octanoylation ((CH2)8-QRFSR) or biotinylation (biotine-[(CH2)6]- QRFSR) led to compounds displaying diminished inhibitory potency towards hap-N. On the other hand, pGlu1-RFSR and [(CH2)8]-QRFSR peptides displayed at least equivalent inhibitory potency for rhNEP compared to opiorphin native peptide.
The importance of the free C-terminal carboxyl group of the QRFSR-COOH peptide in inhibitory potency toward rhNEP, in particular, rhNEP CarboxyDiPeptidase activity. Indeed, the amidation of the C terminal (QRFSR-CONH2) gives rise to a compound displaying diminished inhibitory potency toward rhNEP.
The key role played by the aromatic side chain of Phe1 residue (QRFSR) in the inhibitory potency of opiorphin toward rhNEP and rhAP-N activities. Indeed, substitution with a Tyr residue (QRYSR) led to a compound displaying up to an 8-fold decrease in rhAP-N inhibition potency and a slight decrease in rhNEP inhibition potency. Substitution by an Ala residue led to a compound with completely diminished inhibitory potency toward both rhNEP and rhAP-N [18].
The importance of the RFS central residues of the QRFSR peptide in the inhibitory potency of opiorphin toward rhNEP. The compounds QRGPR – QHNPR – QRFPR displayed equivalent inhibitory potency toward rhAP-N but a low or totally diminished inhibitory potential for rhNEP.
The importance of the guanidium side chains of the Arg2 (R2) and Arg5 (R5) residues in the inhibitory potency of opiorphin toward rhAPN. Indeed, their respective substitution by the ε- amine side chain of Lys residue (QKFSR and QRFSK) led to compounds displaying more than a 10-fold decrease in rhAP-N inhibitory potency while showing equivalent rhNEP inhibitory potency. Their respective substitution by an Ala residue confirmed these results [18].
In summary, there is a clear structural selectivity in the functional interaction of opiorphin with both human NEP and AP-N ectoenkephalinases. The aromatic residue of Phe3 plays a critical role in the interactions of opiorphin with both targets. In addition, the C-terminal FSR tri-amino acids constitute the minimal active sequence for NEP inhibition; moreover, FSR-peptide is 10 times more active than the natural QRFSR-peptide in its inhibition potency toward rhNEP. Conversely, it seems that the entire amino acid sequence of opiorphin is required for full rhAP-N inhibition. In general, our results demonstrate that any change in the intra-peptide sequence inhibits or even abolishes at least one of the two inhibitory activities. In contrast, addition of an amide link with a Tyr residue at the N-terminal position of the peptide ([Y]-QRFSR) does not reduce the inhibitory potency of the peptide toward either human target and does not affect its antinociceptive potency in a pain rat model [1].
Metabolism of the native opiorphin peptide
In order to evaluate the half-life of circulating opiorphin in the human bloodstream, the fate of the natural peptide was analyzed using in vitro kinetic models. The metabolic profile of opiorphin native peptide in human plasma as a function of incubation time at 37°C is shown in Figure 1A. The major metabolism products, generated following a 60-min incubation period of 1 μg QRFSR/QR [3H-F] SR per500 μl human fresh plasma, were isolated by RP-HPLC in conjunction with PDA (224 nm) and radiometric detections and semi-quantified using Chromquest software. The data are expressed by relative peak height. The addition of tracer quantity of tritiated opiorphin established the drug plasma concentration with high precision even for small amounts of compound not usually detected using standard PDA detection. Finally, analyses with Kinetica software were used to predict from the concentration-time course the metabolic half-life of the native compound, either from plasma-induced hydrolysis and/or chemical changes.
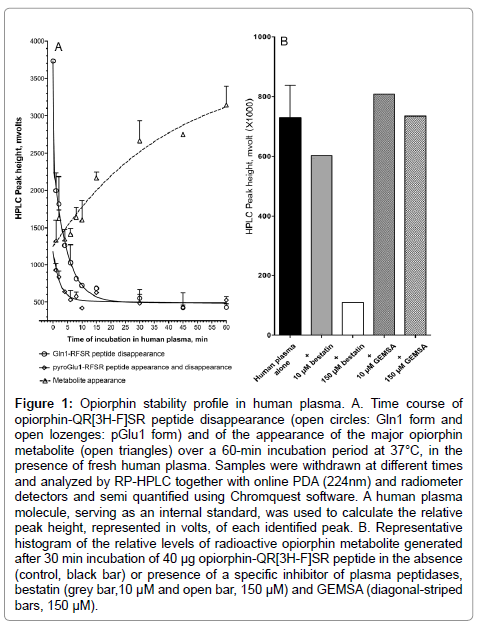
Figure 1: Opiorphin stability profile in human plasma. A. Time course of opiorphin-QR[3H-F]SR peptide disappearance (open circles: Gln1 form and open lozenges: pGlu1 form) and of the appearance of the major opiorphin metabolite (open triangles) over a 60-min incubation period at 37°C, in the presence of fresh human plasma. Samples were withdrawn at different times and analyzed by RP-HPLC together with online PDA (224nm) and radiometer detectors and semi quantified using Chromquest software. A human plasma molecule, serving as an internal standard, was used to calculate the relative peak height, represented in volts, of each identified peak. B. Representative histogram of the relative levels of radioactive opiorphin metabolite generated after 30 min incubation of 40 μg opiorphin-QR[3H-F]SR peptide in the absence (control, black bar) or presence of a specific inhibitor of plasma peptidases, bestatin (grey bar,10 μM and open bar, 150 μM) and GEMSA (diagonal-striped bars, 150 μM).
Native QRFSR-peptide disappears from human plasma with a metabolic half-life evaluated at 5 min (R2=0.88, n=5 time points over the 8 min time course of incubation). One metabolite appeared as early as 1 min after incubation, reaching maximum relative levels after 30 min incubation. Its appearance inversely correlated with the disappearance of Gln1-RFSR native peptide (Figure 1A). The maximal appearance after 30 min incubation was blocked in the presence of 150 μM bestatin, a selective inhibitor of amino peptidases (Figure 1B). In contrast, its appearance was not affected by 150 μM GEMSA, a selective carboxy peptidase B inhibitor (Figure 1B). This result suggests that opiorphinis primarily hydrolyzed to an RFSR-peptide metabolite resulting from the activity of a plasma exo-aminopeptidase, potentially a glutamyl peptidase. Interestingly, the RFSR-peptide is about 3 fold less inhibitory than the native QRFSR-peptide toward both rhNEP and rhAP-N. To increase the 5 minutes half-life of native opiorphin, changes were designed at the level of this sensitive site.
Two additional radioactive molecular populations were distinguished on the RP-HPLC chromatograms during the time course of incubation of QR [3H-F] SR-opiorphin in human plasma. The pGlu1- RFSR-peptide was observed, peaking to a maximum of 16% at 1 min time-point then disappearing with a similar metabolic half-life (T1/2- life=6 min, R2=0.83 for n=4 time points over the 6 min time course of incubation (Figure 1A), to the parent Gln1-RFSR-peptide. This result does not concur with a previous report indicating that pGlu formation (in enzymatic or non-enzymatic processes) minimizes susceptibility to degradation by aminopeptidase [19]. It is also interesting to point out that pGlu1-RFSR peptide is an efficient NEP inhibitor. To a lesser extent, a more hydrophilic molecular population was also observed on the HPLC profile, reaching a maximum from the 2 min time-point and remaining stable at about 12% over the 30 min incubation period. The chromatographic and kinetic behaviors of this population lead us to suggest that it could result from an opiorphin-related product binding to a human plasma component.
Selection of potent bioactive opiorphin peptidomimetics: The peptidomimetic strategy consists of altering the physical characteristics of a peptide without changing its biological activity. Here we wished to design and select functional derivatives of opiorphin that would display in vivo bioavailability properties superior to the native peptide, in particular increased resistance against proteolytic degradation. Several modifications are known to improve the metabolic stability of peptides. Conventional modifications consist of protecting the NH2-and COOH-terminal ends by N-acetylation and C-amidation, respectively. However, SAR studies (see above) reveal that these modifications inhibit or even abolish opiorphin inhibitory potencies. Alternatively, amino acids can be selectively substituted with non-natural amino acids, most notably by a D-enantiomer or β-amino acid [20]. However, as previously reported, ⇓ changes on the structural conformation of N- and C- terminal amino acids (N- and C-terminally homologated opiorphin, ß2hGln-Arg-Phe-Ser-ß3hArg),while increasing by about 7-fold the metabolic half-life of the modified opiorphin in human plasma, reduced by up to 10-fold its inhibitory potency toward both targets. This indicated that the relocation of the terminal carboxy and/ or amino groups has an impact on opiorphin interaction with the enkephalin-inactivating NEP and AP-N [21].
A third possibility to increase the enzymatic stability of peptides is to reduce their peptide character (pseudo peptides), substituting peptide bonds with isosteric surrogates. The isosters most frequently used are the reduced peptide bond (methyl-amino, CH2-NH), the retro-inverso link, the aza group or polyethylene chain spacers such as (CH2-)6- or (CH2-)12-. Depending on the chemical residue in corporated, the most direct consequences are increased resistance to the lytic action of circulating peptidases and an increase in lipophilicity that serves to facilitate transport across biological barriers [20,22]. However, such chemically stabilized peptides can lose some, if not all, of their biological activity, such as the retro-inverso D-amino acid opiorphin analog that lost its ability to inhibit NEP (unpublished observations by Rougeot C).
Consistent with the above, most of the QRFSR-peptide changes failed to reproduce the biological activity of the natural peptide. However, a series of opiorphin derivatives were screened and selected step by step on the basis of their dual inhibitory potency for hNEP and/ or hAP-N. To test for specificity, hit compounds were further tested with respect to other members of the metallo-ectopeptidase family, such as DPPIV and ECE. Here we present only functional opiorphin derivatives displaying significant in vitro inhibitory activity toward human NEP and AP-N.
QRF-[S-O-Octanoyl]-R peptide
Comparative conformational analyses of the opiorphin peptide revealed that the hydroxyl group of the Ser4 residue does not seem to play a critical role in its bioactive conformation for hNEP [18]. Therefore, initially we tested the product resulting from esterification by octanoic acid, [CH2)8], of the serine hydroxyl group of the QRFSR peptide.
As shown in Figure 2, QRF-[S-O-(CH2)8]-R peptide prevented, in a concentration dependent manner, the Abz-dR-G-L-EDDnp cleavage, mediated by recombinant hNEP-Endopeptidase (rhNEP-endo) activity, with a half inhibitory potency (IC50) at 2.1 ± 0.2 μM (r2=0.99, n=30 determination points). It also prevented, in a concentration dependent manner, the Abz-R-G-F-K-DnpOH cleavage, mediated by recombinant hNEP-Carboxy DiPeptidase (rhNEP-CDP) activity, with an IC50 at 13 ± 3 μM (r2=0.98, n=30 determination points) and also the Mca-R-P-P-G-F-S-A-F-K-(Dnp)-OH FRET-peptide (Mca- BK2) cleavage with an IC50 at 14 ± 4 μM (r2=0.99, n=18 determination points). In addition, QRF-[S-O-(CH2)8]-R peptide inhibited, in a dose dependent manner, the Ala-AMC cleavage, mediated by recombinant hAP-N (rhAP-N) activity, with an IC50 at 12 ± 1 μM (r2=0.99, n=27 determination points).
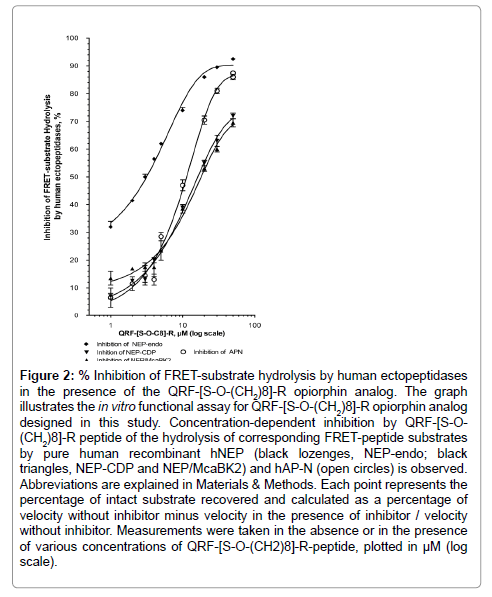
Figure 2: Inhibition of FRET-substrate hydrolysis by human ectopeptidases in the presence of the QRF-[S-O-(CH2)8]-R opiorphin analog. The graph illustrates the in vitro functional assay for QRF-[S-O-(CH2)8]-R opiorphin analog designed in this study. Concentration-dependent inhibition by QRF-[S-O- (CH2)8]-R peptide of the hydrolysis of corresponding FRET-peptide substrates by pure human recombinant hNEP (black lozenges, NEP-endo; black triangles, NEP-CDP and NEP/McaBK2) and hAP-N (open circles) is observed. Abbreviations are explained in Materials & Methods. Each point represents the percentage of intact substrate recovered and calculated as a percentage of velocity without inhibitor minus velocity in the presence of inhibitor / velocity without inhibitor. Measurements were taken in the absence or in the presence of various concentrations of QRF-[S-O-(CH2)8]-R-peptide, plotted in μM (log scale).
QRF[S-O-(CH2)8] R peptide was an essentially equipotent inhibitor compound toward AP-N compared to opiorphin natural peptide. The Ser4 octanoylation of opiorphin-peptide reinforces, by up to 10-fold, the inhibitory potency for rhNEP-Endopeptidase and CarboxyDipeptidase activity, suggesting that the addition of a hydrophobic moiety on the Ser4 side chain induces more favorable contacts with the hydrophobic pocket of the NEP catalytic site.
[C]-QRFSR peptide
We previously showed that addition of a Tyr residue at the N-terminal position of the opiorphin-peptide does not affect its in vitro inhibitory potency or its in vivo antinociceptive properties [1]. Potent NEP and AP-N inhibitors were designed on the basis that the molecules contain a strong metal-coordinating group [23]. These observations were also used to design an opiorphin peptidomimetic carrying at the N-terminal moiety a Cys-thiol functional group that is a strong Zn atom-coordinating group.
As shown in Figure 3, the [C]-QRFSR peptide inhibited, in a concentration dependent manner, the rhNEP-endo activity with an IC50 at 7 ± 1 μM (r2=0.96, n=28 determination points), the rhNEP-CDP with an IC50 at 7 ± 1 μM (r2=0.96, n=31 determination points) and the NEP-dependent Mca-BK2 cleavage with an IC50 at 17 ± 2 μM (r2=0.99, n=16 determination points). It also prevented, in a dose dependent manner, the Ala-AMC cleavage mediated by rhAP-N activity, with an IC50 at 0.8 ± 0.1 μM (r2=0.99, n=30 determination points).
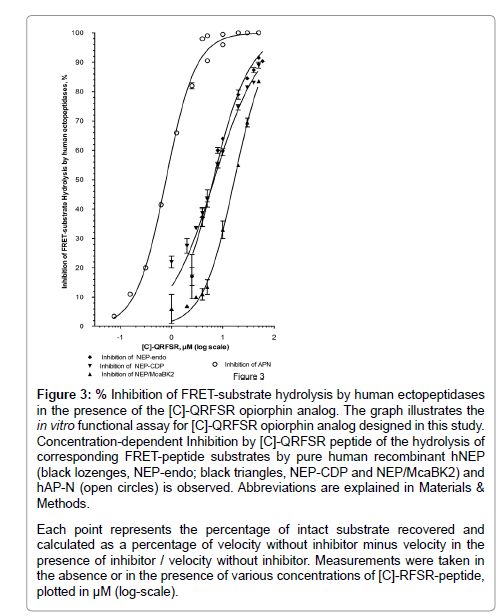
Figure 3: % Inhibition of FRET-substrate hydrolysis by human ectopeptidases in the presence of the [C]-QRFSR opiorphin analog. The graph illustrates the in vitro functional assay for [C]-QRFSR opiorphin analog designed in this study. Concentration-dependent Inhibition by [C]-QRFSR peptide of the hydrolysis of corresponding FRET-peptide substrates by pure human recombinant hNEP (black lozenges, NEP-endo; black triangles, NEP-CDP and NEP/McaBK2) and hAP-N (open circles) is observed. Abbreviations are explained in Materials & Methods.
Each point represents the percentage of intact substrate recovered and calculated as a percentage of velocity without inhibitor minus velocity in the presence of inhibitor / velocity without inhibitor. Measurements were taken in the absence or in the presence of various concentrations of [C]-RFSR-peptide, plotted in μM (log-scale).
Thus, the NH2-[C]-QRFSR-COOH peptide, containing a thiol coordinating group with the Zn atom, displays reinforced inhibitory potency toward hAP-N and hNEP Zn-dependent metalloectopeptidases, 10-fold and 5-fold compared to QRFSR natural peptide, respectively.
[C]-[amino-hexanoic-acid spacer]-QRFSR peptide
In an attempt to protect the opiorphin derivative against degradation by circulating amino peptidases and thus increase its metabolic stability, a [CH2]6 polyethylene bridge [amino-hexanoic-acid spacer] was substituted for the peptide bond joining the Zn-chelating Cys0 and the Glu1 amino acids.
The resulting [C]-[CH2]6-QRFSR peptide showed a dosedependent inhibition of rhNEP- ndo activity with an IC50 at 40 ± 5 μM (r2=0.97, n=24 determination points), of rhNEP-CDP activity with an IC50 evaluated at about 158 μM (r2=0.89, n=25 determination points), and rhAP-N activity with an IC50 at 0.8 ± 0.1 μM (r2=0.99, n=16 determination points).
Surprisingly, the additional polyethylene bridge between the Cys0 and Glu1 residues of the [C]-QRFSR peptide was caused a decrease in inhibitory potency, of more than one order of magnitude relative to [C]-QRFSR peptide, toward NEP enzyme and particularly toward NEP-carboxypeptidase activity, whereas no difference in affinity towards AP-N was detected relative to [C]-QRFSR peptide.
[C]-[amino-hexanoic-acid spacer]-QRF-[S-O-Octanoyl]-R peptide
The [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R derivative, resulting from the combination of the three modifications above, was tested for its inhibitory potency toward rhNEP and rhAP-N. As shown in Figure 4, it inhibited, in a concentration dependent manner, rhNEP-Endo activity with an IC50 at 0.83 ± 0.08 μM (r2=0.99, n=48 determination points), rhNEP-CDP activity with an IC50 at 0.85 ± 0.06 μM (r2=0.99, n=27 determination points) and Mca-BK2 cleavage mediated by the rhNEP with an IC50 at about 2.1 ± 0.1 μM (r2=0.99, n=18 determination points). The [C]-[(CH2)6]-QRF-[S-O-(CH2)6]-R peptide had equipotent inhibitory capacity towards rhAP-N activity with an IC50 at 0.95 ± 0.10 μM (r2=0.99, n=24 determination points). Thus the combination of a well-balanced bioactive profile with equipotent inhibitory capacity encouraged us to retain [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R as the best performing opiorphin peptidomimetic and it was submitted to further exploration.
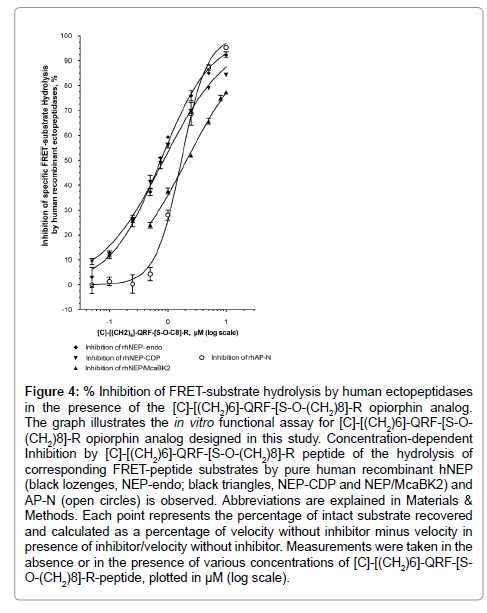
Figure 4: Inhibition of FRET-substrate hydrolysis by human ectopeptidases in the presence of the [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R opiorphin analog. The graph illustrates the in vitro functional assay for [C]-[(CH2)6]-QRF-[S-O- (CH2)8]-R opiorphin analog designed in this study. Concentration-dependent Inhibition by [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R peptide of the hydrolysis of corresponding FRET-peptide substrates by pure human recombinant hNEP (black lozenges, NEP-endo; black triangles, NEP-CDP and NEP/McaBK2) and AP-N (open circles) is observed. Abbreviations are explained in Materials & Methods. Each point represents the percentage of intact substrate recovered and calculated as a percentage of velocity without inhibitor minus velocity in presence of inhibitor/velocity without inhibitor. Measurements were taken in the absence or in the presence of various concentrations of [C]-[(CH2)6]-QRF-[SO-( CH2)8]-R-peptide, plotted in μM (log scale).
In the biologically relevant in vitro assay, using substance P, the physiological NEP substrate and human cell membranes as sources of native human NEP, the [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R peptide prevented, in a concentration dependent manner, substance P cleavage mediated by membrane-bound hNEP-Endopeptidase (mhNEP-Endo) activity with an IC50 at 1.6 ± 0.4 μM (r2=0.95, n=13 determination points) (Figure 5). Under the same assay conditions, it appears to be at least five times more potent than opiorphin natural peptide toward hNEP [1]. In addition using fluorescent substrates with human cell membranes as sources of native hNEP, the [C]-[(CH2)6]- QRF-[S-O-(CH2)8]-R peptide inhibited in a concentration dependent manner the mhNEP-Endo activity with an IC50 at 1.6 ± 0.4 μM, and mhAP-N activity with an IC50 at 0.9 ± 0.1 μM (Figure 5). Thus, the designed analog presents similar affinity towards human NEP and APN, whether they are in a native membrane-anchored or recombinant soluble conformation.
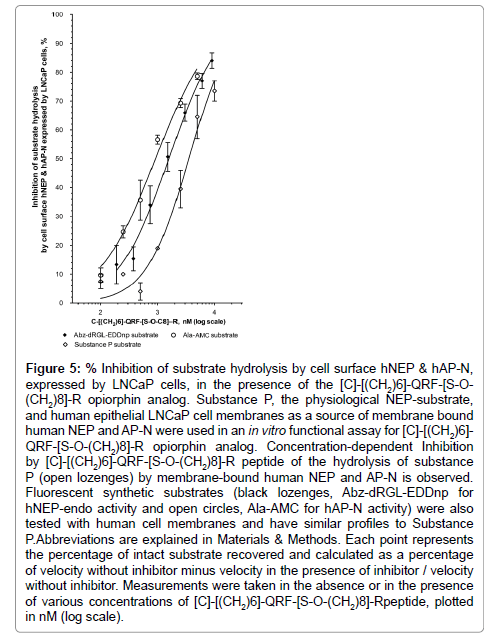
Figure 5: Inhibition of substrate hydrolysis by cell surface hNEP & hAP-N, expressed by LNCaP cells, in the presence of the [C]-[(CH2)6]-QRF-[S-O- (CH2)8]-R opiorphin analog. Substance P, the physiological NEP-substrate, and human epithelial LNCaP cell membranes as a source of membrane bound human NEP and AP-N were used in an in vitro functional assay for [C]-[(CH2)6]- QRF-[S-O-(CH2)8]-R opiorphin analog. Concentration-dependent Inhibition by [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R peptide of the hydrolysis of substance P (open lozenges) by membrane-bound human NEP and AP-N is observed. Fluorescent synthetic substrates (black lozenges, Abz-dRGL-EDDnp for hNEP-endo activity and open circles, Ala-AMC for hAP-N activity) were also tested with human cell membranes and have similar profiles to Substance P.Abbreviations are explained in Materials & Methods. Each point represents the percentage of intact substrate recovered and calculated as a percentage of velocity without inhibitor minus velocity in the presence of inhibitor / velocity without inhibitor. Measurements were taken in the absence or in the presence of various concentrations of [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-Rpeptide, plotted in nM (log scale).
in vitro assays using human recombinant DPP4 or ECE-1 revealed that the [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R compound did not inhibit rhECE1 and rhDPPIV-ectopeptidase activities even at 100 μM final concentration. These results indicate that, similarly to opiorphin, the opiorphin analog shows excellent selectivity with respect to related zinc-metallo peptidases, such as ECE1 (closely structurally related to NEP with 40% sequence identity) and DPPIV that is involved among other endopeptidases, including NEP, in the inactivation of the substance Pandbradykinin.
Di-peptide analogs can be metabolically more resistant to peptidase degradation. We tested the cystine-dipeptide (single disulfide bond connecting the Cys1 residue of each peptide chain) of [C]-[(CH2)6]- QRF-[S-O-(CH2)8]-R for its inhibitory potency towards human NEP and AP-N. The resulting [C-[(CH2)6]-QRF-[S-O-(CH2)8]-R] 2cystine-dipeptideanalog showed a dose dependent inhibition of the rhNEP-Endo activity with an IC50 at 0.99 ± 0.07 μM (r2=0.99, n=16 determination points), and of the rhAP-N activity with an IC50 at 0.58 ± 0.05 μM (r2=0.99, n=15 determination points). Thus, in vitro the dipeptide demonstrates similar inhibitory potency towards hNEP and hAP-N when compared to the monomer.
[dCys]-QRF-[Ser-O-octanoyl]-[dArg]
Another strategy to protect peptide compounds against degradation by circulating peptidases is the replacement of the N-term and C-term amino acid residues, which are major targets for degradation by circulating exopeptidases, by their respective D-enantiomer.
As shown in Figure 6, [dC]-QRF-[S-O-(CH2)8]-[dR] derivative peptide inhibited, in a concentration dependent manner, rhNEPEndo activity with an IC50 at 4 ± 1 μM (r2=0.97, n=30 determination points) and rhNEP-CDP activity with an IC50 at 21 ± 1 μM (r2=0.99, n=30 determination points). Strikingly, this derivative was at least 200 times more potent against rhAP-N activity than against rhNEP with an IC50 at 0.022 ± 0.002 μM (r2=0.98, n=43 determination points). We also used human cell membranes as a source of native membrane-bound hNEP and hAP-N and confirmed that the [dC]-QRF-[S-O-(CH2)8]- [dR] peptide displays an unbalanced inhibitory profile. Indeed, it showed a dose-dependent inhibition of mhNEP-Endo activity with an IC50 at 9 ± 1 μM (r2=0.98, n=21 determination points) and of mhNEPCDP activity with an IC50 at 37 ± 5 μM (r2=0.95, n=21 determination points). In addition, it appeared to be 30-100 times more potent toward mhAP-N activity than toward mhNEP(IC50 at 0.3 ± 0.1 μM, r2=0.95, n=21 determination points).
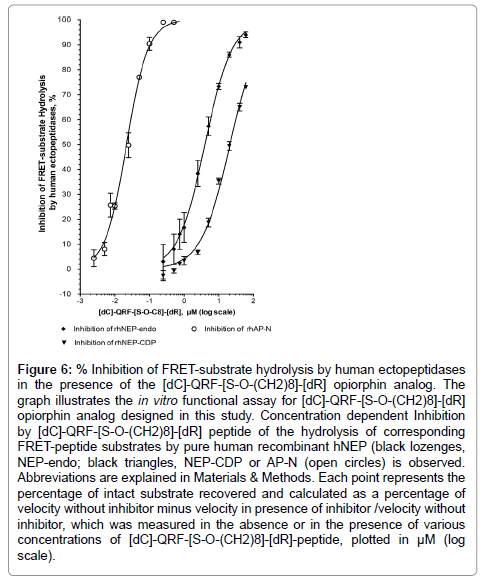
Figure 6: % Inhibition of FRET-substrate hydrolysis by human ectopeptidases in the presence of the [dC]-QRF-[S-O-(CH2)8]-[dR] opiorphin analog. The graph illustrates the in vitro functional assay for [dC]-QRF-[S-O-(CH2)8]-[dR] opiorphin analog designed in this study. Concentration dependent Inhibition by [dC]-QRF-[S-O-(CH2)8]-[dR] peptide of the hydrolysis of corresponding FRET-peptide substrates by pure human recombinant hNEP (black lozenges, NEP-endo; black triangles, NEP-CDP or AP-N (open circles) is observed. Abbreviations are explained in Materials & Methods. Each point represents the percentage of intact substrate recovered and calculated as a percentage of velocity without inhibitor minus velocity in presence of inhibitor /velocity without inhibitor, which was measured in the absence or in the presence of various concentrations of [dC]-QRF-[S-O-(CH2)8]-[dR]-peptide, plotted in μM (log scale).
Furthermore, substitution of the L-Arg5 by its respective D-enantiomer clearly affected the inhibitory potency of the compound toward hNEP-carboxydipeptidase. The related [dC]-QRF-[S-O- (CH2)8]-R peptide inhibited mhNEP-CDP activity with an IC50 at 2.6 ± 0.3 μM (r2=0.98, n=30 determination points), about ten times more potent than the D-Arg5 counterpart. Such a difference leads us to propose the existence of a stereo-chemical requirement for optimal interaction of the peptide with the catalytic site of NEP. Conversely, the substitution of the L-Cys0 by its respective D-enantiomer clearly enhanced the inhibitory potency of the compound toward hAP-N (about 50 times more potent than the L-Cys0 counterpart) and may be due to the fact that its spatial conformation provides tight binding to the AP-N target.
The [dC]-QRF-[S-O-(CH2)8]-[dR] derivative probably displays some superior in vivo bioavailability properties compared to native opiorphin peptide, such as a possible gain in circulating amino- and carboxy -peptidase resistance. However, it’s very modest gain in hNEP inhibitory potency, combined with a distinctly unbalanced bioactive profile eliminated it as a suitable candidate molecule. Therefore, only the C-[(CH2)6]-QRF-[S-O-(CH2)8]-R derivative was retained for further exploration.
Metabolism and Toxicity profile of the best performing opiorphin functional derivative
Metabolism in fresh human plasma: We established overall in vitro pharmacokinetic and metabolic parameters, based on an in vitro time-dependent system, using opiorphin or its derivative incubated in human plasma. Kinetica software, which is used to predict the metabolic half-life (T½) of the parent peptide from the concentrationtime course, was used in this study.
As shown above, in vitro kinetic analyses in human plasma revealed that the native QRFSR-peptide disappears with a half-life evaluated at 5 min. Its disappearance results in part from the cyclization of Gln1 (16% maximum) but mainly from the hydrolytic removal of both Gln1- and pGlu1-peptides by plasma amino peptidases (reaching a maximum of 84% of the parent peptide at 60 min incubation) and also, to a small extent (12%), from potential complex formation.
The in vitro kinetic change experiments in human plasma of the [C]-[(CH2)6]-QRF[S-O-(CH2)8]-R derivative, were done with 50 μg peptide/500 μl human plasma by RP-HPLC relative quantification and ELISA quantitative assay. The concentration-time profile was analyzed by Prism (Figure 7) and Kinetica softwares. The half-time disappearance of [C]-[(CH2)6]-QRF[S-O-(CH2)8]-R was evaluated at 4.5 min (R2=0.89, n=5 time points over 5 min time course of incubation). Its disappearance results primarily from the dimerization (36% maximum of [[C]-[(CH2)6]-QRF[S-O-(CH2)8]-R]2-dimer which has a T½ disappearance of 41 min; R2=0.87, n=7 time points over the 60 min time course of incubation). ELISA and HPLC analyses showed that 40% of the parent peptide remains stable in the human plasma even after 60 min incubation (14% for the QRFSR-peptide), indicating that the [C]-[(CH2)6]-QRF[S-O-(CH2)8]-R derivative is more metabolically stable in human plasma than opiorphin native peptide. In addition, it is important to point out that the major biotransformation product of the parent derivative, the cystine-dipeptide, is as active as the parent peptide in fluorescence-based NEP and AP-N assays.
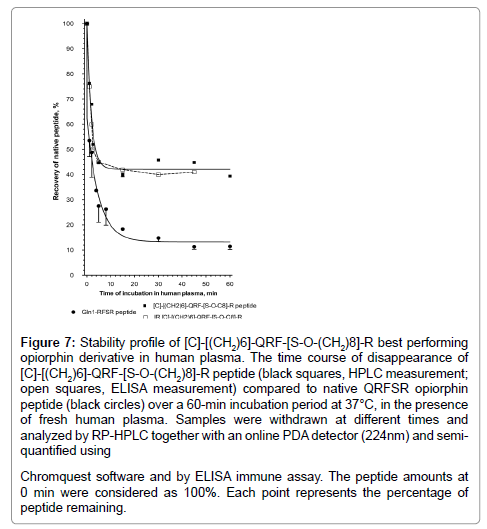
Figure 7: Stability profile of [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R best performing opiorphin derivative in human plasma. The time course of disappearance of [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R peptide (black squares, HPLC measurement; open squares, ELISA measurement) compared to native QRFSR opiorphin peptide (black circles) over a 60-min incubation period at 37°C, in the presence of fresh human plasma. Samples were withdrawn at different times and analyzed by RP-HPLC together with an online PDA detector (224nm) and semiquantified using Chromquest software and by ELISA immune assay. The peptide amounts at 0 min were considered as 100%. Each point represents the percentage of peptide remaining.
All together, the data showed that, as expected, opiorphin derivative [C]-[(CH2)6-spacer]-QRF[S-O-(CH2)8]-R demonstrates much greater metabolic stability with respect to human plasma aminopeptidases compared to the native opiorphin peptide.
Drug Absorption and in vitro Cytotoxicity: A range of in vitro ADME-Tox assays provided by Cerep Laboratories (Celle L’Evescault- France) allowed us to evaluate a number of factors including drug absorption and membrane permeability with the A-B permeability and P-glycoprotein ATPase efflux system [24]. The Caco-2/TC7 (pH 6.5/7.4) human cell line gives an indication of the intestinal epithelial transport potential of compounds [24]. Metabolic stability, using human liver microsomes and in vitro cytotoxicity in cell-based assays that measure cellular parameters such as cell viability, nuclear size and mitochondrial membrane potential using the HepG2 human cell line can also be evaluated.
Our data demonstrate that, compared to the reference positive and negative controls, no apparent in vitro human cell toxicityis observed for either QRFSR native peptide or [C]-[(CH2)6]-QRF-[SO-( CH2)8]-R derivative peptide, incubated at 10, 30 and 100 μM final concentrations for 72 h at 37°C. For example, relative to controls at 100 μM, the peptides increased cell proliferation by 1and 12%, respectively and reduced nuclear size and mitochondrial membrane potential only by 1 and 5%, respectively. However, there is a clear decrease in the metabolic stability of the opiorphin derivative in the presence of human liver microsomes compared with the native opiorphin peptide: at 10 μM final concentration and after 60 min incubation, 2.5% of the parent [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R compound remains versus 47% remaining in the case of opiorphin. Surprisingly, the opiorphin derivative, although endowed with higher lipophilicity than opiorphin native peptide, did not display significantly increased trans-membrane cell permeability over the 60 min incubation-period at 37°C, as the apparent permeability coefficient of both tested compounds was <0.2×10-6 cm/s (10 μM test concentration and HPLC-MS/MS detection method). However, this result is probably due to the cellular model used, namely, TC7 human epithelial intestinal cells derived from the CaCO2 cell line, and known to express membrane-bound NEP and AP-N ectoenzymes. The cell line, therefore, is not an appropriate model for permeability studies of NEP and/or AP-N- inhibitor-ligands. Indeed, the mean recovery of the compounds in donor samples was dramatically low (0% for QRFSR and 14% for the derivative) due mainly to binding to TC7 cell membranes.
We then tested in vivo acute toxicity using a rat model provided by CERB (Centre de Recherches Biologiques, Baugy-France). CERB experimental conditions are based on a stepwise procedure, each step uses 3 male rats for each compound. No mortality occurred among the animals treated with QRFSR natural peptide at 100 mg/kg maximum dose, administered as a bolus in the caudal vein. This dose is a 100-fold the effective I.V. dose in the rat pain model. In contrast, the rat treated with a 100 mg/kg dose of [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R analog died 3 minutes after treatment; however, no mortality occurred among the 3 animals treated at 30 mg/kg I.V. These animals were further observed for general clinical and neurobehavioral signs, based on the Irwin method, for 14 days [25]. No clinical signs were observed during the course of the study of both peptides. Body weight gain was normal and no gross organ or tissue changes were detected by necropsy.
In conclusion, under the experimental conditions adopted by CERB, opiorphin natural QRFSR peptide administered intravenously at 100 mg/kg and [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R derivative peptide administered at 30 mg/kg did not induce signs of toxicity.
Antinociceptive effect in the rat formalin pain model: The exciting in vitro results from FRET-based NEP and AP-N assays prompted us to study the analgesic efficacy of the [C]-[(CH2)6]-QRF- [S-O-(CH2)8]-R analog, in terms of delay of action-both potency and duration of action, in a rat model.
Using the behavioral formalin-induced pain model, known as the Formalin Test, we investigated the anti-nociceptive potency of the [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R analog. The duration of formalininjected paw licking (sec) and the total number of inflamed paw flinches and body tremors were recorded over the 60 min-test period. The formalin test measures the behavioral response to a chemical-induced inflammatory nociception, which induces two distinct nociceptive phases separated a stationary interphase: a early acute phase (first 10 min after formalin injection) followed by a late phase in which a more tonic pain is elicited.
Here, we demonstrate that the [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R functional opiorphin analog inhibits, in a dose-dependent manner, the pain behavior induced by long-acting chemical stimuli with significant antinociceptive effect at 0.5, 1 and 2 mg/kg I.V. doses over early and later phases of the test (Figure 8). Thus, compared to the control vehicle rats, the opiorphin analog-treated rats at 1 and 2 mg/kg dose spent significantly less time in paw licking over the first 10 min-test period, from 161 ± 19 sec (vehicle) to 89 ± 15 sec (1 mg/kg) and 77 ± 6 sec (2 mg/kg) (P<0.05 and 0.01 vs vehicle by Mann-Whitney U-test, MWT, n=8 rats/group) as morphine-treated rats at 2 mg/kg I.V. dose (43 ± 13 sec, P<0.01 by MWT). The 1 and 2 mg-treated rats also spent significant less time in paw licking over the second 10-30 min period, from 468 ± 48 sec (vehicle) to 287 ± 28 sec (1 mg/kg) and 224 ± 27 sec (2 mg/kg) (P<0.01 and 0.001 vs vehicle by MWT, n=8 rats/group). The 0.5 mg/kg-treated rats also spent at least 30% less time in inflamed paw licking over pain periods: 98 ± 10 sec (early phase) and 334 ± 54 sec (late phase) compared to vehicle-treated rats 161 ± 19 sec and 468 ± 48 sec, respectively (P<0.05 vs vehicle by MWT, n=8 rats/group). From 30 min post-formalin injection, the duration of paw licking decreased in a parallel manner in both vehicle-and opiorphin analog-treated rats and their behavioral responses to the test compound as well as to morphine were not significant. Conversely, during this 30-60 min period, the control vehicle rats exhibited an important increase in the total number of formalin-injected paw flinches and body tremors. And systemic administration of opiorphin analog at 2 mg/kg significantly reduced this pain behavioral score throughout the 30 to 60 min timeperiod, from 300 ± 31 (vehicle) to 218 ± 15 (P ≤ 0.05 vs vehicle by MWT, n=8 rats/group).
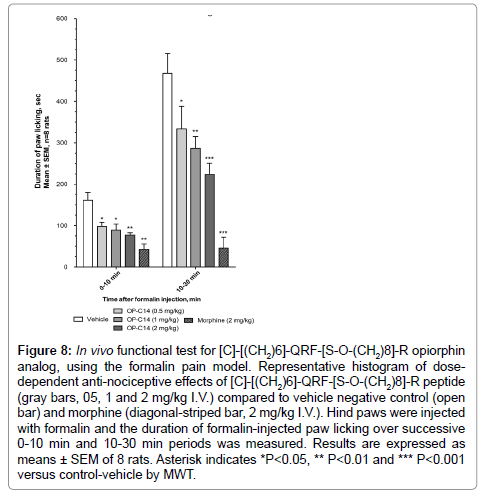
Figure 8: In vivo functional test for [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R opiorphin analog, using the formalin pain model. Representative histogram of dosedependent anti-nociceptive effects of [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R peptide (gray bars, 05, 1 and 2 mg/kg I.V.) compared to vehicle negative control (open bar) and morphine (diagonal-striped bar, 2 mg/kg I.V.). Hind paws were injected with formalin and the duration of formalin-injected paw licking over successive 0-10 min and 10-30 min periods was measured. Results are expressed as means ± SEM of 8 rats. Asterisk indicates *P<0.05, ** P<0.01 and *** P<0.001 versus control-vehicle by MWT.
This model was previously used for testing native opiorphin activity and we demonstrated that opiorphin, at 1 and 2 mg/kg I.V. doses inhibits nociception in both acute early and tonic late phases of the test by primarily activating μ-opioid pathways [1,10].
Conversely, under the same experimental conditions, we observed that the [C]-[(CH2)6]-QRF-[S-O-(CH2)8]-R cystine-dipeptide failed to significantly inhibit pain behavioral responses at 1 and 2 mg/kg tested doses (data not shown).
Thus, our data clearly indicate that the [C]-[(CH2)6]-QRF-[S-O- (CH2)8]-R opiorphin analog inhibits nociception induced by acute and long-acting chemical stimuli in the rat model. Strikingly, although metabolically more resistant and more potent in its ability to inhibit enkephalin-degrading ectopeptidases, the opiorphin analog-induced pain reduction in the formalin test is similar to the opiorphin natural peptide, in terms of dose effect, delay and duration of action. This could be due to the loss of a significant proportion of active derivative by dimerization and/or by hepatic metabolism in vivo in rats.
The goal of the study described here was to design and characterize functional analogs of opiorphin that display in vivo bioavailability properties superior to the native peptide. The inhibitory potency of the main functional derivatives toward human NEP and AP-N is summarized in Table 1. A close structural selectivity in the functional interaction of opiorphin with both human NEP and AP-N targets was first demonstrated by SAR studies, thus limiting the possibilities of chemical changes. Nevertheless, results of the study clearly demonstrate that addition of a N-terminal Zn-chelating group, a Cys-thiol group and replacement of the first labile peptide bond by a polyethylene surrogate, a [CH2]6 linker, and, finally, substitution of Ser4 by a octanoyl-Ser, Ser-O-[CH2]8, to the native opiorphin amino acid sequence produced a high performing C-[(CH2)6]-QRF[S-O-[CH2]8]-R derivative. This designed analog displays reinforced inhibitory potency toward hAP-N activity (more than 10-fold increase) and toward hNEPEndopeptidase and CarboxyDiPeptidase activities (more than 40-fold increase) relative to the QRFSR natural peptide. Moreover, the analog shows increased stability in human plasma compared to unmodified opiorphin. Finally, we demonstrate that it retains the full analgesic activity characteristic of the opiorphin native peptide, in terms of delay of action and effective doses, in the behavioral formalin-induced pain rat model. If we consider that the maximum effective analgesic dose for the two compounds is 1 mg/kg I.V., the safety-effectiveness ratio is estimated at 30 for the designed analog and at 100 for the native peptide.
| Opiorphin & derivatives | IC50, µM toward hAP-N |
IC50, µM toward hNEP- Endopeptidase | IC50, µM toward hNEP- CarboxyDiPeptidase |
|---|---|---|---|
| Native opiorphin QRFSR | 10 ± 2 (rAPN) | 46 ± 5 (rNEP) 29 ± 3 (rmNEP) 11 ± 3 (mNEP/SP) |
57 ± 4 (rNEP) 33 ± 6 (rmNEP) |
| QRF-[S-O-(CH2)8]-R | 12 ± 1 | 2.1 ± 0.2 | 13 ± 3 |
| [C]-QRFSR | 0.8 ± 0.1 | 7 ± 1 | 7 ± 1 |
| [C]-[(CH2)6]-QRFSR | 0.8 ± 0.1 | 40 ± 5 | # 158 |
| C-[(CH2)6]-QRF-[S-O-(CH2)8]-R | 0.9 ± 0.1 (rAPN) 0.9 ± 0.1 (mAPN) |
0.8 ± 0.1 (rNEP) 1.6 ± 0.4 (mNEP) 1.6 ± 0.4 (mNEP/SP) |
0.9 ± 0.1 (rNEP) |
| [dC]-QRF-[S-O-(CH2)8]-[dR] | 0.022 ± 0.002 (rAPN) 0.3 ± 0.1 (mAPN) |
4 ± 1 (rNEP) 9 ± 1 (mNEP) |
21 ± 1 (rNEP) 37 ± 5 (mNEP) |
Table 1: Comparative inhibition potencies of Opiorphin and peptidomimetic compounds toward human NEP and AP-N. Compound concentration for 50% inhibition of hNEP and hAP-N activity is expressed as mean ± SEM of at least 13 determination points. rAP-N/rNEP: soluble recombinant ectoenzymes; mAP-N/mNEP: membrane-bound native ectoenzymes expressed by LNCaP epithelial cells; rmAP-N/rmNEP: membrane-bound recombinant ectoenzymes expressed by transfected HEK cells. Numbers in bold highlight increased inhibitory potency relative to the native peptide.
C-[(CH2)6]-QRF[S-O-[CH2]8]-R, has improved opiorphin pharmacological parameters and thus could be a promising analgesic drug-candidatein a preclinical and clinical setting.
This work was in large part supported by funding sources from the “Direction de la Valorisation et des Partenariats Industriels” and “Dons et Mécénat d'entreprises” Institut Pasteur. The maunscript was edited by traduction@lefevere-laoide.net
The present address of A. Bogeas: Team Glial Plasticity, U894 Inserm, Université Paris Descartes, Paris, France
No conflicts of interest, financial or otherwise, are declared by the author(s).