Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Research Article - (2018) Volume 7, Issue 1
The stereoselective total synthesis of the proposed structure of naturally occurring nonenolide stagonolide D has been achieved using D-mannitol as the starting material. The latter has been utilized for the preparation of both the olefinic alcohol segment and the olefinic acid segment of the target molecule. The synthetic sequence involves asymmetric epoxidation and ring-closing metathesis as the key steps.
Keywords: Stagonolide D, D-mannitol, Stereoselective synthesis, Asymmetric epoxidation, Ring-closing metathesis
Nonenolides (ten membered lactonic compounds) are common naturally occurring secondary metabolites. Stagonolides are recent examples of these compounds [1-4]. Various stagonolides have been isolated from liquid and solid cultures of Stagonospora cirsii, a fungal pathogen obtained from Cirsium arvense [1-4]. Some of these natural products have been examined to exhibit antibacterial, antifungal and phytotoxic properties [1-4]. However, as the stagonolides are naturally available in scarce amounts, detailed investigation on their biological properties is difficult. The term “proposed” is used because different other structures are also possible with the given spectral data of the molecule. However, earlier methods used different synthetic path ways stating from expensive starting materials [5,6]. Herein, we report a simple and efficient approach to the total synthesis of stagonolide D (1), starting from the inexpensive and easily available starting material, D-mannitol.
In continuation of our work on the stereoselective synthesis of natural products we have accomplished the total synthesis of the proposed structure of stagonolide D (1) [3]. The purposed of this study is to make an efficient synthesis of Stagonolide D and related molecules. The prepared compounds can be utilized for their bio-evolution.
The retrosynthetic analysis of 1 revealed that it’s both the two core fragments, the olefinic alcohol 2 and the olefinic acid 3 can be generated from D-mannitol 4 (Scheme 1).
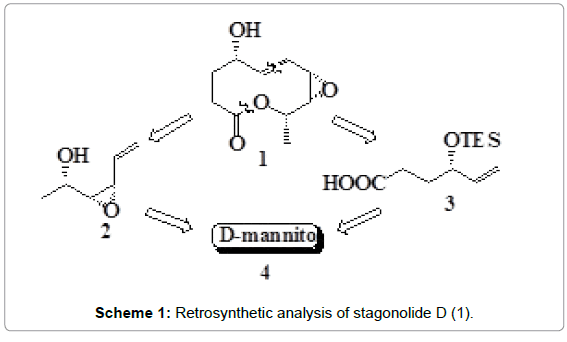
Scheme 1: Retrosynthetic analysis of stagonolide D (1).
The present synthesis of the fragment 2 was initiated (Scheme 2) by converting D-mannitol 4 into the olefin 5 by reported method [7]. Compound 5 was reacted with PivCl in the presence of Et3N and DMAP in CH2Cl2 when its primary hydroxyl group was protected to furnish the product 6 in high yield.
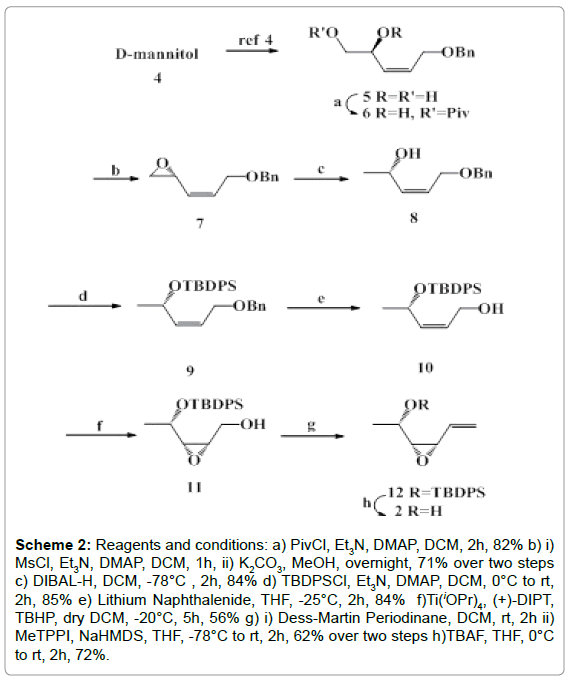
Scheme 2: Reagents and conditions: a) PivCl, Et3N, DMAP, DCM, 2h, 82% b) i) MsCl, Et3N, DMAP, DCM, 1h, ii) K2CO3, MeOH, overnight, 71% over two steps c) DIBAL-H, DCM, -78°C , 2h, 84% d) TBDPSCl, Et3N, DMAP, DCM, 0°C to rt, 2h, 85% e) Lithium Naphthalenide, THF, -25°C, 2h, 84% f)Ti(iOPr)4, (+)-DIPT, TBHP, dry DCM, -20°C, 5h, 56% g) i) Dess-Martin Periodinane, DCM, rt, 2h ii) MeTPPI, NaHMDS, THF, -78°C to rt, 2h, 62% over two steps h)TBAF, THF, 0°C to rt, 2h, 72%.
Treatment of 6 with MsCl using Et3N and DMAP followed by reaction with methanolic KOH afforded the α-epoxide 7 [8]. Reduction of this compound 7 with DIBAL-H produced the compound 8 having α-OH group. The free hydroxyl group of 8 was protected as TBDPS ether to form 9 which on subsequent hydrogenation over Lithium Naphthalenide generated the epoxy alcohol 10. Compound 10 was utilized for sharpless asymmetric epoxidation [9-11] using Ti(Oipr)4, (+)-DIPT and TBHP in CH2Cl2 at -20°C to yield the epoxy alcohol 11 which was purified for further steps. Compound 11 underwent oxidation with Dess Martin periodinane (DMP) to the corresponding aldehyde which on C-1 Wittig olefination with triphenyl phosphonium methyl iodide yielded the olefinic epoxide 12. The TBDPS ether group of 12 was deprotected with TBAF in THF to furnish the desired olefinic alcohol 2.
The second fragment 3 was also prepared from D-mannitol 4 (Schemes 3 and 4) by converting it into the diol 13 by reported method [12]. Both the hydroxyl groups of this diol 13 were protected by reacting it with TESCl in the presence of DMAP in CH2Cl2 to afford the compound 14. The primary TES ether group of 14 was then selectively oxidised [13] under Swern conditions and the resulting aldehyde underwent C-1 Wittig olefination by treatment with triphenyl phosphonium methyl iodide to give the olefinic compound 15 (Scheme 2).
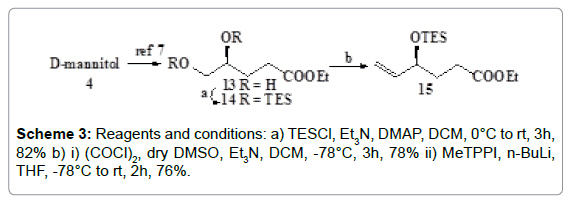
Scheme 3: Reagents and conditions: a) TESCl, Et3N, DMAP, DCM, 0°C to rt, 3h, 82% b) i) (COCl)2, dry DMSO, Et3N, DCM, -78°C, 3h, 78% ii) MeTPPI, n-BuLi, THF, -78°C to rt, 2h, 76%.
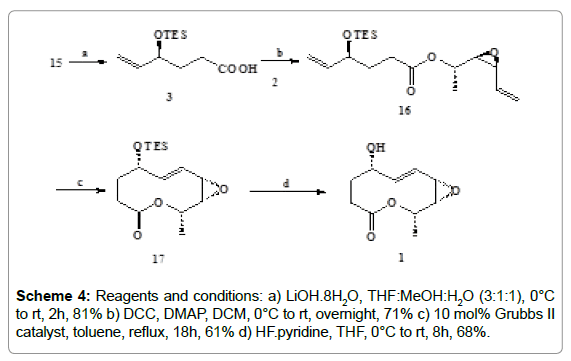
Scheme 4: Reagents and conditions: a) LiOH.8H2O, THF:MeOH:H2O (3:1:1), 0°C to rt, 2h, 81% b) DCC, DMAP, DCM, 0°C to rt, overnight, 71% c) 10 mol% Grubbs II catalyst, toluene, reflux, 18h, 61% d) HF.pyridine, THF, 0°C to rt, 8h, 68%.
The ester group of 15 was hydrolysed (Scheme 4) with LiOH. 8H2O in THF-MeOH-H2O (3:1:1) to yield the olefinic acid 3 (not isolated) which was subsequently coupled with olefinic alcohol 2 in the presence of DCC and DMAP in CH2Cl2 (Scheme 3).
The resulting diene 16 was then subjected to ring closing metathesis (RCM) using Grubbs’ second generation catalyst [14-16] to produce the nonenolide 17 which was purified by column chromatography. Finally the deprotection of the TES ether group of 17 with HF-pyridine in THF afforded stagonolide D. We could not get proper crystals of the compound required for X-ray crystallographic analysis. The physical and spectral properties of this compound was found to be identical to those reported for the natural product (having proposed structure of stagonolide D) of earlier synthesis (Scheme 4) [11].
General experimental procedure
The silica gel F254 plates were used for thin layer chromatography (TLC) in which the spots were examined under UV light and then developed by an iodine vapor. Column chromatography was performed with silica gel (BDH 100-200 mesh). Solvents were purified according to standard procedures. The spectra were recorded with the following instruments; IR: Perkin-Elmer RX FT-IR spectrophotometer; NMR: Varian Gemini 200 MHz (1H) and 50 MHz (13C) spectrometer; ESIMS: VG-Autospec micromass. Organic extracts were dried over anhydrous Na2SO4. Optical rotations were measured with JASCO DIP 300 digital polarimeter at 25°.
(S, Z)-5-(benzyloxy)-2-Hydroxypent-3-enyl pivalate (6): To a stirred solution of 5 (4.5 g, 21.6 mM) in dry CH2Cl2 (30 mL), Et3N (4.50 mL, 32.4 mM), was added. After 5 min PivCl (2.64 mL, 21.6 mM) and catalytic amount of DMAP (260 mg, 2.16 mM) were added and stirred at room temperature for 2 h. The solvent mixture was evaporated and concentrated under reduced pressure. The residue was purified by column chromatography to afford compound 6 (5.18 g, 82% yield) as a colorless liquid. IR: 3448, 2926, 1631 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.38-7.20 (5H, m), 5.78 (1H, m), 5.52 (1H, m), 4.61 (1H, m), 4.52 (2H, s), 4.18-3.94 (3H, m), 3.58 (1H, m), 2.10 (9H, s); 13C NMR (75 MHz, CDCl3); δ 179.1, 138.0, 131.0, 130.6, 128.7, 128.0, 73.2, 67.6, 66.8, 66.0, 39.0, 31.8, 22.4; ESIMS: m/z 315 [M+Na]+. Anal. Cald. for C17H24O4: C, 69.84; H, 8.27; O, 21.89%; Found: C, 69.81; H, 8.25; O, 21.91%.
(R, Z)-2-(3-(benzyloxy)prop-1-enyl)oxirane (7): To a stirred solution of 6 (5 g, 17.12 mM) in dry CH2Cl2 (25 mL), Et3N (4.50 mL, 32.4 mM), was added. After 5 min MsCl (1.59 mL, 20.5 mM) and catalytic amount of DMAP were added and stirred at room temperature for 1 h. The solvent was evaporated and the residue (5.57 g, 88%) was used directly for further reaction. To a stirred solution of the residue (5.3 g, 14.32 mM) in dry MeOH (30 mL) at room temperature, K2CO3 (3.9 g, 28.64 mM) was added and stirred for overnight. The reaction mixture was filtered, the filtrate was washed with sat. NaHCO3 (3 × 40 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography to afford the chiral epoxide 7 (2.04 g, 75%). [α]D25=+20.22 (c=0.225, CHCl3); IR: 1653, 1458, 1077 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.45-7.28 (5H, m), 5.91 (1H, m), 5.22 (1H, t, J=7.0 Hz), 4.58 (2H, s), 4.25 (2H, d, J=7.0 Hz), 3.60 (1H, m), 2.99 (1H, m), 2.62 (1H, m); 13C NMR (75 MHz, CDCl3); δ 138.1, 132.2, 132.0, 130.4, 128.2, 127.9, 72.1, 65.3, 48.8, 48.0; ESIMS: m/z 213 [M+Na]+. Anal. Cald. for C12H14O2: C, 75.76; H, 7.42; O, 16.82%; Found: C, 75.80; H, 7.40; O, 16.80%.
(S, Z)-5-(benzyloxy)pent-3-en-2-ol (8): To a stirred solution of chiral epoxide compound 7 (1.9 g, 10 mM) in DCM (60 mL) DIBAL-H 1.44 M in hexane (32.9 mL) was added drop wise at -78°C. The mixture was stirred at 0°C for another 1 h. After completion of the reaction, excess DIBAL-H was quenched with 10% Roche’s salt solution and extracted with DCM (2 × 50 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography to obtain 8 (1.61 g, 84%) as clear oil. IR: 3445, 2930, 2858, 1647, 1108, 1078 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.39- 7.22 (5H, m), 5.84 (1H, m), 5.65 (1H, m), 4.53 (2H, s), 4.31 (1H, m), 4.08 (2H, d, J=10.0 Hz), 1.28 (3H, d, J=7.0 Hz); 13C NMR (75 MHz, CDCl3); δ 138.2, 130.2, 129.3, 128.8, 128.6, 127.5, 72.5, 65.2, 62.1, 29.9; ESIMS: m/z 215 [M+Na]+. Anal. Cald. for C12H16O2: C, 74.97; H, 8.39; O, 16.64%; Found: C, 74.80; H, 8.40; O, 16.62%.
(S, Z)-(5-(benzyloxy)pent-3-en-2-yloxy)(tert-butyl)diphenylsilane (9): To a stirred solution of 8 (1.5 g, 7.81 mM) in dry CH2Cl2 (30 mL), Et3N (2.71 mL, 19.5 mM) was added. After 5 min TBDPSCl (2.8mL, 10.9 mM) and catalytic amount of DMAP (95 mg, 0.781 mM) were added and stirred at room temperature for 2 h. The solvent mixture was evaporated and the residue was purified by column chromatography to afford 9 (2.85 g, 85% yield) as a colorless liquid. IR: 2932, 2858, 1468, 1427, 1108 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.76-7.58 (4H, m), 7.51-7.28 (6H, m), 7.39-7.23 (5H, m), 5.82 (1H, m), 5.65 (1H, m), 4.53 (2H, s), 4.34 (1H, m), 4.08 (2H, d, J=7.0 Hz), 1.28 (3H, d, J=7.0 Hz), 1.05 (9H, s); 13C NMR (75 MHz, CDCl3); δ 137.5, 134.2, 132.6, 130.2, 130.0, 128.8, 128.6, 75.0, 69.1, 63.2, 26.8, 24.5, 19.4; ESIMS: m/z 453 [M+Na]+. Anal. Cald. for C28H34O2Si: C, 78.09; H, 7.96; O, 7.43%; Found: C, 78.06; H, 7.97; O, 7.41%.
(S, Z)-4-(tert-butyldiphenylsilyloxy)pent-2-en-1-ol (10): To a solution of naphthalene (1.35 g, 4.8 mM) in THF (25 mL), were added lithium metal (70 mg, 4.6 mM) in small pieces. The reaction mixture was stirred at room temperature under inert conditions until lithium metal was completely dissolved (nearly 3 h). The resulting dark green solution of lithium naphthalenide was then cooled to -25°C, followed by addition of a solution of 9 (2.6 g, 6.04 mM) in THF (15 mL) dropwise over 5 min. The resulting mixture was stirred at -25°C for 70 min. Later, the reaction mixture was quenched with NH4Cl (10 mL) and concentrated in vacuo. The crude residue so obtained was purified by column chromatography to afford alcohol 10 (1.54 g, 75% yield) as a brown color liquid. IR: 3449, 2930, 2858, 1427, 1108 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.74-7.57 (4H, m), 7.51-7.34 (6H, m), 5.96 (1H, m), 5.82 (1H, m), 4.34 (2H, d, J=7.0 Hz), 3.98 (1H, m), 1.46 (3H, d, J=7.0 Hz), 1.05 (9H, s); 13C NMR (75 MHz, CDCl3); δ 134.4, 132.6, 130.2, 130.0, 129.7, 128.6, 61.3, 58.9, 25.5, 24.6, 19.5; ESIMS: m/z 363 [M+Na]+. Anal. Cald. for C21H28O2Si: C, 74.07; H, 8.29; O, 9.40%; Found: C, 74.09; H, 8.27; O, 9.41%.
((2S,3S)-3-((S)-1-(tert-butyldiphenylsilyloxy)ethyl)oxiran-2-yl) methanol (11): In a 100 mL two-necked round-bottomed flask, 10 mL of dry CH2Cl2 was added to 4Å powdered activated molecular sieves and suspension mixture was cooled to -25°C, Ti(OiPr)4 (0.6 mL, 2.05 mM) and (+) DIPT (0.6 mL, 2.87 mM) in dry CH2Cl2 (10 mL) were added subsequently with stirring and the resulting mixture was stirred for 30 min at -25°C. Then compound 10 (1.4 g, 4.11 mM) in dry CH2Cl2 (20 mL), TBHP (0.6 g, 1.88 mM) were added and the resulting mixture was stirred for 5 h at -25°C. It was then warmed to 0°C, quenched with 1 mL of water, and stirred for 1 h at rt. After that 30% aqueous NaOH solution saturated with NaCl (1 mL) was added and the reaction mixture was stirred vigorously for another 30 min at rt. The resulting mixture was then filtered through Celite rinsing with CH2Cl2. The organic phase was separated and the aqueous phase was extracted with CH2Cl2. Combined organic phases were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and purified by silica gel column chromatography to afford 11 (0.82 g, 56%) as a viscous liquid. IR: 3449, 2930, 2858, 1632, 1427, 1219, 1108 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.76-7.57 (4H, m), 7.51-7.28 (6H, m), 3.78-3.50 (2H, m), 3.42 (1H, m), 2.91 (1H, m), 2.65 (1H, m), 1.28 (3H, d, J=7.0 Hz), 1.05 (9H, s); 13C NMR (75 MHz, CDCl3); δ 136.0, 133.9, 130.1, 127.5, 68.8, 61.2, 58.7, 57.3, 27.0, 21.2, 19.6; ESIMS: m/z 379 [M+Na]+. Anal. Cald. for C21H28O3Si: C, 70.74; H, 7.92; O, 13.46%; Found: C, 70.76; H, 7.90; O, 13.39%.
Tert-butyldiphenyl((S)-1-((2S,3S)-3-vinyloxiran-2-yl)ethoxy) silane (12): To a stirred solution of 11 (790 mg, 2.22 mM) in dry CH2Cl2 (20 mL), NaHCO3 (372 mg, 4.44 mM) was added at 0°C. Dess-Martin Periodinane (1.88 g, 4.44 mM) was added and the reaction mixture was stirred at rt for 2 h. The reaction mixture was quenched with a saturated solution of NaHCO3 and sodium thiosulfate (1:7) (5 mL) at 0°C and extracted with CH2Cl2 (3 × 10 mL). The organic layer was washed with water (10 mL) and dried (Na2SO4). The solvent was evaporated and the residue (aldehyde 707 mg, 90%) was used directly for further reaction. To a stirred solution of above aldehyde in dry THF (25 mL), NaHMDS (3.7 mL, 3.68 mM), MeTPPI were added and stirred for 2 h at 0°C to rt. Solvent was removed under reduced pressure and extracted with EtOAc (2 × 5 mL), the combined organic layers were washed with brine (5 mL), and dried (Na2SO4). The solvent was evaporated and the residue was purified by column chromatography to afford compound 12 (387 mg, 60% yield) as a colorless liquid. [α]D 25=-18.1 (c=1.25, CHCl3); IR: 3030, 2923, 2855, 1454, 1359, 1246, 1106 cm-1; 1H NMR (300 MHz, CDCl3): δ 7.75-7.66 (4H, m), 7.48-7.37 (6H, m), 5.65 (1H, m), 5.37- 5.26 (2H, m), 3.42 (1H, t, J=7.0 Hz), 3.18 (1H, m), 2.94 (1H, m), 1.22 (3H, d, J=7.0 Hz), 1.05 (9H, s); 13C NMR (75 MHz, CDCl3); δ 135.3, 133.0, 129.8, 127.7, 118.1, 68.5, 61.8, 57.2, 27.2, 21.1, 19.0; ESIMS: m/z 375 [M+Na]+. Anal. Cald. for C22H28O2Si: C, 75.76; H, 7.42; O, 16.82%; Found: C, 75.80; H, 7.40; O, 16.80%.
(S)-1-((2R,3S)-3-vinyloxiran-2-yl)ethanol (2): To a stirred solution of compound 12 (350 mg, 0.99 mM) in dry THF (30 mL) at 0°C TBAF (0.316 mL, 1.09 mM) was added and stirred at room temperature for 2 h. The reaction mixture was evaporated and the residue was purified by column chromatography to afford olefinic alcohol fragment 2 (81 mg, 72% yield) as a colorless liquid. IR: 3423, 1467, 1428, 1371, 1108, 1036 cm-1; 1H NMR (300 MHz, CDCl3): δ 5.60 (1H, m), 5.39-5.30 (2H, m), 3.40 (1H, m), 3.16 (1H, m), 2.92 (1H, m), 1.69 (1H, brs), 1.22 (3H, d, J=7.0Hz); 13C NMR (75 MHz, CDCl3); δ 135.2, 119.3, 69.5, 60.8, 58.2, 20.4; EIMS: m/z 114 [M]+. Anal. Cald. for C6H10O2: C, 63.14; H, 8.83; O, 28.03%; Found: C, 63.17; H, 8.81; O, 28.05%.
In conclusion, we have accomplished stereoselective total synthesis of the proposed structure of stagonolide D starting from easily available D-mannitol and involving asymmetric epoxidation and ring-closing metathesis as the key steps.
The author thanks CSIR-UGC, New Delhi for financial assistance.