Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Opinion - (2014) Volume 3, Issue 3
During early human development, females randomly inactivate one of the two X chromosomes in a process called X chromosome inactivation. Since this process is usually random, most women are a balanced 50:50 mosaic of cells. While this is the situation in young females, mosaicism is usually lost in the elderly, particularly in the blood, a process referred to as skewing. We have previously shown, using primary cell populations, that skewing is highly predictable in vitro and results from hemizygous selection whereby a competitive advantage is conferred to all cells that express genes from one X chromosome. Here we examine the mechanism behind skewing in vitro and conclude that the major factor is X-linked polymorphism. Similar observations are made in vivo where we find that most adult female mice skew in a predictable pattern towards a dominant X. These findings have important implications for in vitro studies and offer a platform to gain insights into the dynamics of skewing in the hematopoietic system.
<Since the process that inactivates X chromosomes in humans is random [1,2], females are a mosaic of cells, expressing genes either from the maternal or the paternal X in a balanced ratio of 50:50 [2]. A deviation of ≥ 25% from balanced inactivation of each parental X chromosome is defined as skewing [3,4]. Skewing results either through nonrandom choice of X chromosome inactivation during early developmental stages (primary skewing) or through secondary selection in favor of cells expressing a particular X chromosome (referred to as secondary skewing). In the mouse, the Xce locus influences the choice of which X will undergo inactivation during early embryonic stages. By contrast, humans do not carry an Xce locus and young women are usually a balanced mosaic of cells. The observation that secondary, age-related skewing is significantly more common in elderly females [5-8] suggests that skewing in humans is mainly caused by secondary events that take place during adult life.
One theory is that age-related secondary skewing results from genetic drift in stem cells, i.e. loss of mosaicism by chance, rather than fitness differences, due to a limited population size. Since the stem cell pool in certain tissues becomes depleted with age this may explain why skewing is more common in the elderly. A drawback of this model however, is that it does not explain the high correlation of the X-specificity in skewing between elderly monozygotic twin pairs [7,9,10], or the correlation in the direction of skewing across tissues obtained from the same individual [11-15].
Secondary skewing may result from hemizygous selection which is defined as a competitive advantage to all cells that express genes from one X chromosome, as a consequence of either negative (e.g. premature senescence of an unfavorable population) and/or positive (e.g. neoplastic clone) selection. Blood cells have a limited lifespan and must be replenished continuously throughout life from a small reserve of hematopoietic stem cells in the bone marrow. Studies conducted in murine genetic models defective in DNA repair, intracellular reactive oxygen species management, and telomere maintenance indicate that all these pathways are critical to the longevity and stress response of the stem cell pool. Since many X-linked genes participate in these pathways, genetic/epigenetic differences between the two X chromosomes may potentially give a survival advantage to one cell population expressing genes from a dominant X allele. Alternatively, skewing may result from a neoplastic clone, as a consequence of chromosomal anomalies that confer a proliferative advantage. This theory may explain the correlation between skewing and the probability of developing several devastating diseases including cancer [2,16-20]. In agreement, many of the anomalies seen in elderly skewed females are characteristic of those found in hematological cancers [19].
While most skewing experiments have been performed in vivo, very few studies have examined the dynamics between the 2 isogenic populations in culture conditions. We have previously shown that when a balanced mosaic population of human female fibroblasts is serially passaged in vitro, cells expressing genes from an unfavorable X chromosome enter senescence at an earlier stage and show more DNA damage than the sibling population [21]. This disadvantage leads to a gradual shift from a balanced ratio of cells toward a completely skewed population homogenously expressing the same X chromosome. Here we further investigate why two cell populations that are cultured under the same conditions and are functionally, morphologically, and genetically (but not epigenetically) identical behave so differently and conclude that these differences stem from X-linked polymorphism. Finally, we extended these experiments to the mouse system both in vitro and in vivo. We first show that as in humans, skewing in vitro occurs in mouse primary somatic cells, and then in a 12 month cohort study, we demonstrate that similar to the situation in vitro, age-related skewing in the murine blood is highly predictable. These findings may give us insights into the dynamics of the hematopoietic system during aging and the correlation between loss of mosaicism and several life threatening conditions.
Skewing in primary cultures results from Hemizigous selection
X chromosome mosaicism was examined in 27 cultured human female fibroblast populations (HFF). While at early passage, all cultures were a balanced mosaic of cells expressing genes either from the maternal or paternal X chromosome, all cultures lost mosaicism within 4 to 12 weeks. Consistent with previous observations [21], for each HFF line, all sister dishes skewed towards a population expressing genes from a dominant X allele (Xd) (Figure S7A). Using HFF in which the 2 sibling populations can be visually distinguished (Figure S1A) we measured the dynamics of X chromosome inactivation upon serial passaging. In all experiments (n=4 independent experiments in sister dishes) cultures lost mosaicism at day 68 (± 3) (Figure S1B,C), indicating that not only Xd but also the kinetics of skewing are highly predictable. These observations point to fundamental differences between the 2 isogenic cell populations.
To gain insights into the mechanism that leads to skewing we isolated sibling populations from mosaic cultures. Early passage HFF cultures derived from 4 donors were cloned by limiting dilution. Generally, clones that express genes from Xd showed better growth potential and produced more clones than their isogenic siblings (Figure S2B). However, these differences were not statistically significant for all lines (Figure S2A), probably due to the small number of clones obtained per line as a result of clonal exhaustion and clone-to-clone variability. Nevertheless, this inconsistency highlights the possibility that skewing may result from an elite subpopulation that is not represented in cloned cultures.
Following cell isolation from tissues, primary cultures may contain small numbers of precursor cell types [22]. Daughter cells of such precursors are expected to maintain their parental X chromosome inactivation pattern and may have a proliferative advantage over other cultured cells, leading to skewing if enough cell divisions are allowed. In such a scenario, tissue cultures derived from different tissues/locations will not necessarily skew synchronously towards the same direction (i.e. each sample may have its own unique elite population, expressing genes from the paternal X in some samples and from the maternal X in others (Figure S7C). To test the ‘elite’ model, we obtained skin samples from two non-related 16 week old female fetuses. Heterogeneous X-linked SNPs were identified (Table S1). For each fetus, HFF were extracted independently from 5 skin samples taken from different locations and further passaged in parallel in sister dishes (Figure 1A). As expected, the cultures were a balanced mosaic at early passages. For each donor all independent cultures consistently skewed towards the same Xd upon serial passaging (Figure 1B), eliminating the involvement of an elite population during skewing in vitro.
| Sample | SNP identity | Variants | Restriction enzyme | Gene | Remarks |
|---|---|---|---|---|---|
| GM00081 | rs894271 | CA[A/G/ T]TTG | Mefl | XIST | father of GM03066 |
| GM04957 | n16992442 | AGCG[C/T]T | AfeI | XIST | father of GM04959 |
| GM00800 | n16992442 | AGCG[C/ T]T | Afel | XIST | father of GM00798 |
| GM07255 | rs16992442 | AGCG[C/ T]T | Afel | XIST | father of G11401494 |
| GM05165 | rs492933 | CA[G/A]TTG | Mefl | OPHN1 | father of GM05167 |
| GM03066 | rs1194271 | CA[A/G/T]TTG | Mefl | XIST | daughter of GM00081 |
| GM04959 | n16992442 | AGCG[C/T]T | Afel | XIST | daughter of GM04957 |
| GM00798 | rs16992442 | AGCG[C/T]T | Afel | XIST | daughter of GM00800 |
| GM01494 | rs16992442 | AGCG[C/T]T | Afel | XIST | daughter of GM07255 |
| Gh105167 | rs492933 | CA[G/A]TTG | Mefl | OPHN1 | daughter of GM05165 |
| Human embryo 1 | rs492933 | CA[G/A]TTG | Mefl | OPHN1 | fibroblasts from skin |
| Human embryo 2 | rs55856360 | [C/ T] TGCAG | Pstl | PDHA | fibroblasts from skin |
| 11738 | rs492933 | CA[G/A]TTG | Mefl | OPHN1 | GM06814 |
| control-3 | rs492933 | CA[G/A]TTG | Mefl | OPHN1 | fibroblasts frim skin |
| Fis 80 | mutation | TCA[C /T]GGT | N.A | MeCP2 | from Ren Syndrom donor |
| control 1 | rs3269 | AAA[G/C]TAA | N.A | Tetraspanin | mother of control-2 |
| control 2 | rs2805901 | GCA[C/T]GGT | N.A | ATPase | daughter of control-1 |
| GM03045 | rs894271 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM03046 | rs894272 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM14439 | rs894273 | CA[A/G/T]TTG | Mefl | XIST | transformed cell int |
| GM03045 | rs894274 | CA[A/G/T]TTG | Mefl | XIST | transformed cell line |
| GM03046 | rs894275 | CA[A/G/T]TTG | Mefl | XIST | transformed cell line |
| GM14439 | rs894276 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM14440 | rs894277 | CA[A/G/T]TTG | Mefl | XIST | transformed cell line |
| GM14447 | rs894278 | CA[A/G/T]TTG | Mefl | XIST | transformed cel line |
| GM14448 | rs894279 | CA[A/G/T]TTG | Mefl | XIST | transformed cell fine |
| GM14464 | rs894280 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM14465 | rs894281 | CA[A/G/T]TTG | Mefl | XIST | transformed col fine |
| GM14476 | rs894282 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM14477 | rs894283 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM14503 | rs894284 | CA[A/G/T]TTG | Mefl | XIST | transformed cell tine |
| GM14504 | rs894285 | CA[A/G/T]TTG | Mefl | XIST | transformed col line |
| GM14535 | rs894286 | CA[A/G/T]TTG | Mefl | XIST | transformed cel line |
| GM14536 | rs894287 | CA[A/G/T]TTG | Mefl | XIST | transformed cell line |
| Mice in vivo | rs214261465 | TTG[C/G]CAA | N.A | IL2RG | crossing CAST with B6 |
Table S1: Cell list. Human cells from anonymous non affected females were a generous gift from Dr. Bruno Reversade, Institute of Medical Biology, Singapore (controls-1,2,3) and Dr. Jerry Chan, National University of Singapore (Human embryos-1,2). The rest of the human cells were purchased from Coriell Cell Repositories. Mice cells were a generous gift from Dr. Barbara Knowls, Institute of Medical Biology, Singapore.
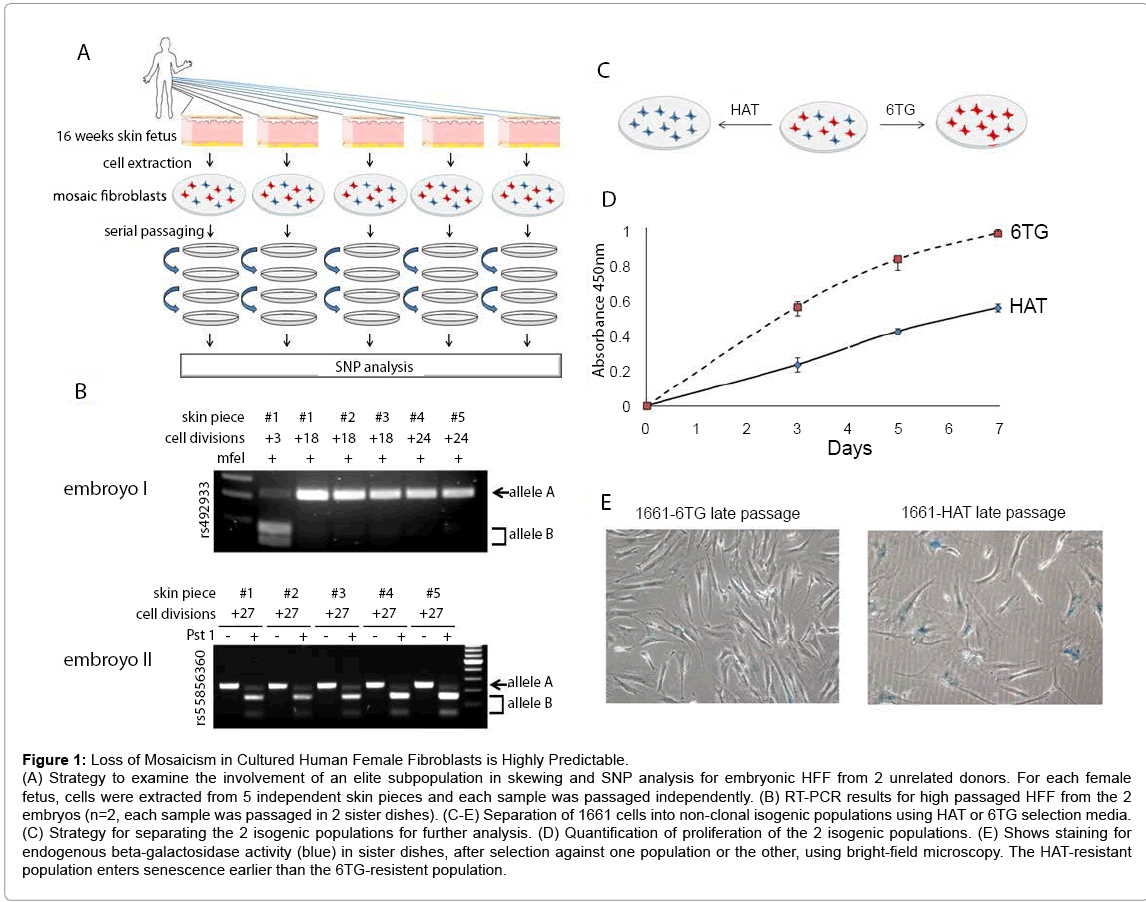
Figure 1: Loss of Mosaicism in Cultured Human Female Fibroblasts is Highly Predictable.
(A) Strategy to examine the involvement of an elite subpopulation in skewing and SNP analysis for embryonic HFF from 2 unrelated donors. For each female fetus, cells were extracted from 5 independent skin pieces and each sample was passaged independently. (B) RT-PCR results for high passaged HFF from the 2 embryos (n=2, each sample was passaged in 2 sister dishes). (C-E) Separation of 1661 cells into non-clonal isogenic populations using HAT or 6TG selection media. (C) Strategy for separating the 2 isogenic populations for further analysis. (D) Quantification of proliferation of the 2 isogenic populations. (E) Shows staining for endogenous beta-galactosidase activity (blue) in sister dishes, after selection against one population or the other, using bright-field microscopy. The HAT-resistant population enters senescence earlier than the 6TG-resistent population.
To demonstrate that skewing results from hemizygous selection, we aimed to compare growth potential between 2 non-clonal sibling populations. To address this, we purchased primary HFF from a Lesch- Nyhan carrier. These cells (HFF GM01661) originate from a Lesch- Nyhan carrier (Table S1) and carry a heterozygous mutation in the X-linked gene HPRT. While cells expressing the non-functional HPRT allele are resistant to the toxic nucleotide analog 6-thio-guanine (6TG), cells that express the functional HPRT allele can grow in the presence of hypoxanthine, aminopterin, and thymidine (HAT)-containing medium. Thus, this biochemical distinction can be used to separate mosaic cultures into two sibling populations (Figure 1C). At early passage, cells grew in either 6TG or HAT, which selects against one population or the other after 6 days (Figure S3A). This indicates that the culture is a mosaic and therefore suggests that the mutation does not affect mosaicism in the donor. On the other hand, high-passage non-treated cultures were resistant to 6TG, but not to HAT medium (Figure S3B), indicating that the Xd is carrying the mutant HPRT gene. In agreement, low passage cultures that are resistant to 6TG and showed a growth advantage (n=3) (Figure 1D), were reprogrammed into iPSCs at a higher efficiency (Figure S3C), and entered senescence later than their siblings, as indicated by beta-galactosidase activity assay (n=2) (Figure 1E). Together, these results suggest that skewing results because cells expressing genes from an unfavorable X allele enter senescence earlier, leading to nonrandom skewing.
To further examine the correlation between skewing and premature senescence of an unfavorable sibling population, we examined cell lines that escaped the Hayflick limit. Fourteen EBV-transformed lymphoblastoid female cell lines were purchased and heterogeneous SNPs were identified (GM03045, GM03046, GM14439, GM14440, GM14447, GM14448, GM14464, GM14465, GM14476, GM14477, GM14503, GM14504, GM14535, GM14536; table S1). As expected, all lines were a balanced mosaic at early passages. However, in contrast to primary cell lines which usually skew in less than 50 days in culture, mosaicism was highly stable. With one exception which skewed after 150 days (not shown) all immortalized lines stayed balanced for at least 5 months in culture (Figure S4). These observations suggest that skewing is less common in transformed cells (Figure 4B) and are consistent with our previous findings that ectopic expression of telomerase alleviates skewing (Figure 4C) (21).
Skewing in Female fibroblasts does not result from X-Linked Imprinted genes
Several studies in adult mice imply that imprinted X-linked genes may play a role in nonrandom skewing in specific tissues [23,24]. If skewing in HFF is influenced by X-linked imprinted genes, Xd is expected to originate from the same parent in all independent cultures (Figure S7D). To examine this we purchased HFF of young females and their parents. The X chromosome inactivation patterns of the daughters were determined by identification of heterogeneous X-linked SNPs (Table S1). Consistent with previous results, sister cultures from each daughter skewed towards the same Xd (not shown). However, while Xd for some daughter was the maternal, the dominant X in others was the paternal allele (Figure 2A). These results argue against the involvement of X-linked imprinted genes in skewing in cultured HFF.
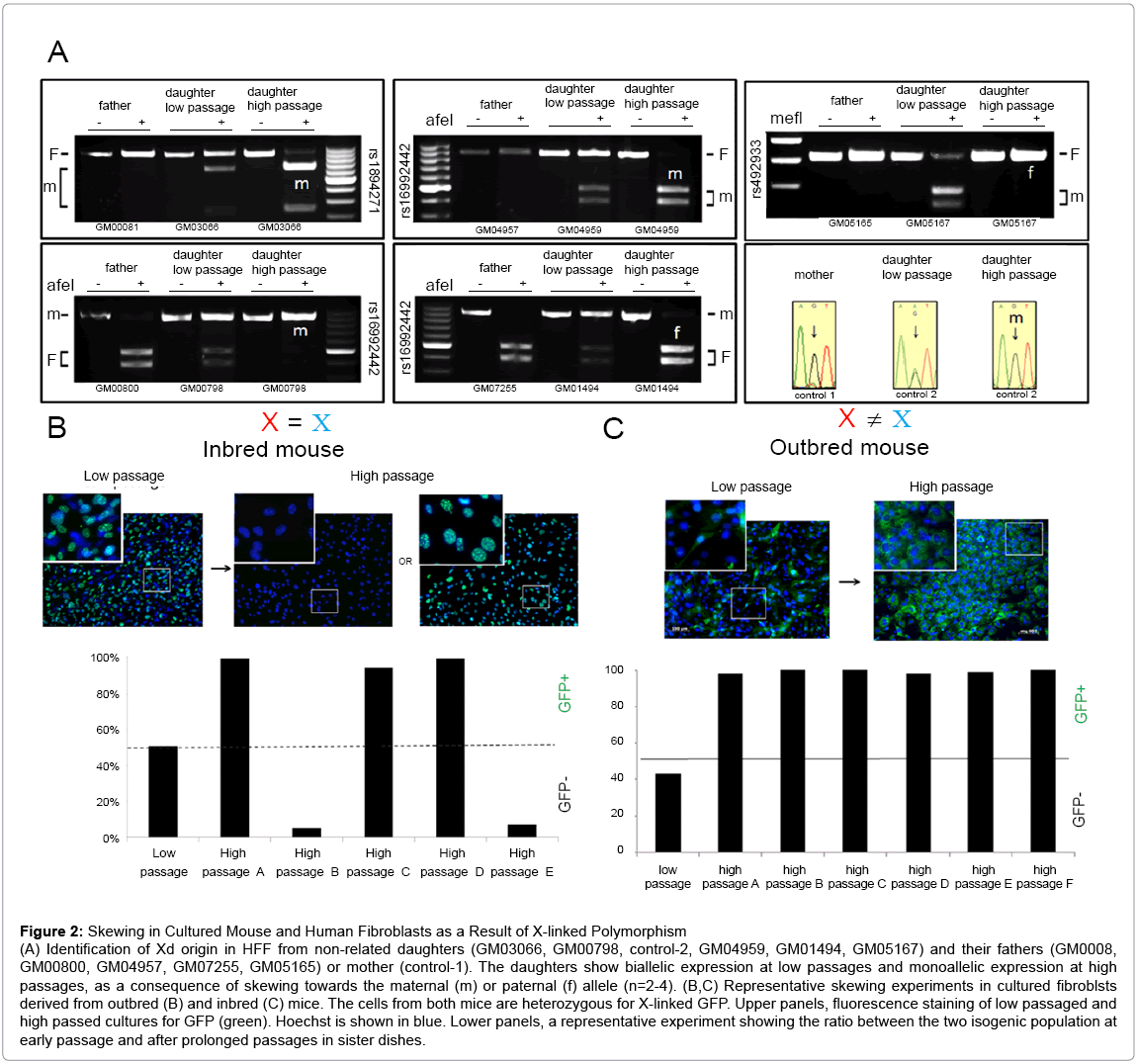
Figure 2: Skewing in Cultured Mouse and Human Fibroblasts as a Result of X-linked Polymorphism
(A) Identification of Xd origin in HFF from non-related daughters (GM03066, GM00798, control-2, GM04959, GM01494, GM05167) and their fathers (GM0008, GM00800, GM04957, GM07255, GM05165) or mother (control-1). The daughters show biallelic expression at low passages and monoallelic expression at high passages, as a consequence of skewing towards the maternal (m) or paternal (f) allele (n=2-4). (B,C) Representative skewing experiments in cultured fibroblsts derived from outbred (B) and inbred (C) mice. The cells from both mice are heterozygous for X-linked GFP. Upper panels, fluorescence staining of low passaged and high passed cultures for GFP (green). Hoechst is shown in blue. Lower panels, a representative experiment showing the ratio between the two isogenic population at early passage and after prolonged passages in sister dishes.
Skewing as a result of X-linked polymorphism
In contrast to inbred mice, humans are highly outbred and therefore our 2 X alleles differ significantly. In agreement, SNP array analysis for sub-clonal populations from 4 independent donors revealed 16.5-42% variability between the two X alleles (not shown). Some of these genetic variations may influence protein function when they fall within coding regions or gene regulation when they fall within promoter/enhancer sequences or snRNA. Since many X-linked genes are involved in important cellular functions, X-linked genetic variants that influence gene expression may lead to skewing. Since inbred-mice (i-mice) carry 2 identical X alleles they offer a unique system to examine the correlation between X-linked polymorphism and skewing. Fibroblasts were extracted from i-mice that carry a GFP construct on one of their X alleles. At early passage, all sister dishes were a balanced 1:1 mosaic of GFP-positive and GFP-negative cells. However, in contrast to human cells, high passage i-mice sister-cultures did not have a preferred Xd allele, and skewed randomly towards one X or the other (n=5) (Figure 2B, 4D). Real-time RTPCR analysis of high passage sister dishes showed similar genomic GFP regardless of the direction of skewing (Figure S5A), indicating that the skewed cultures did not lose an X chromosome during serial passaging. To test the mechanism behind random skewing in i-mice we compared balanced low-passage and skewed high-passage i-mice cultures. Skewed i-mice cultures showed significantly better proliferation potential and faster metabolism than earlier pre-skewed cultures (Figure S5 B,C) suggesting that skewing in cultured i-mice fibroblasts may have resulted from a clonal expansion.
The inconsistency between i-mouse fibroblasts and HFF may result from either species differences or due to a lack of X-linked polymorphism in the mouse. To test this we performed similar experiments using fibroblasts from E12.5 female outbred-mice (o-mice) carrying a GFP construct on one of their X alleles. At early passage, all sister dishes were balanced 1:1 mosaic. After serial passaging in vitro, all o-mice sister dishes consistently skewed towards the same Xd (n=3) (Figure. 2C), indicating that similar to humans, o-mice female fibroblasts lose their mosaic pattern in a consistent direction (Figure 4E). Together, these results suggest that non-random skewing of o-mouse female fibroblasts in vitro results from X-linked polymorphism.
Age-related skewing in vivo is not random
To examine the dynamics of skewing in vivo we reciprocally crossed 4 pairs of C57BL/6 (B6) and CAST/EiJ (CAST) mice. As expected, all 11 female offspring showed some degree of skewing towards the CAST allele presumably as a consequence of heterogeneous Xce locus expression [25] which leads to nonrandom X chromosome inactivation during early developmental stages [25,26-30]. To examine whether skewing continues during adult life, blood was sampled in female mice 2-4 weeks after birth and every 1-3 sequential months for 12 months. Out of 18 mice, 6 animals showed significant additional skewing and 5 animals showed mild additional skewing during adult life (Figure 3 and S6). To the best of our knowledge, this is the first report that demonstrates age-related skewing in adult mice. Strikingly, all 11 mice skewed towards the CAST strain regardless of its parental origin, indicating that similar to primary skewing, the choice of Xd during adult life is not random (Figure 4F). These findings may give us insights regarding to the dynamics of the hematopoietic system during aging.
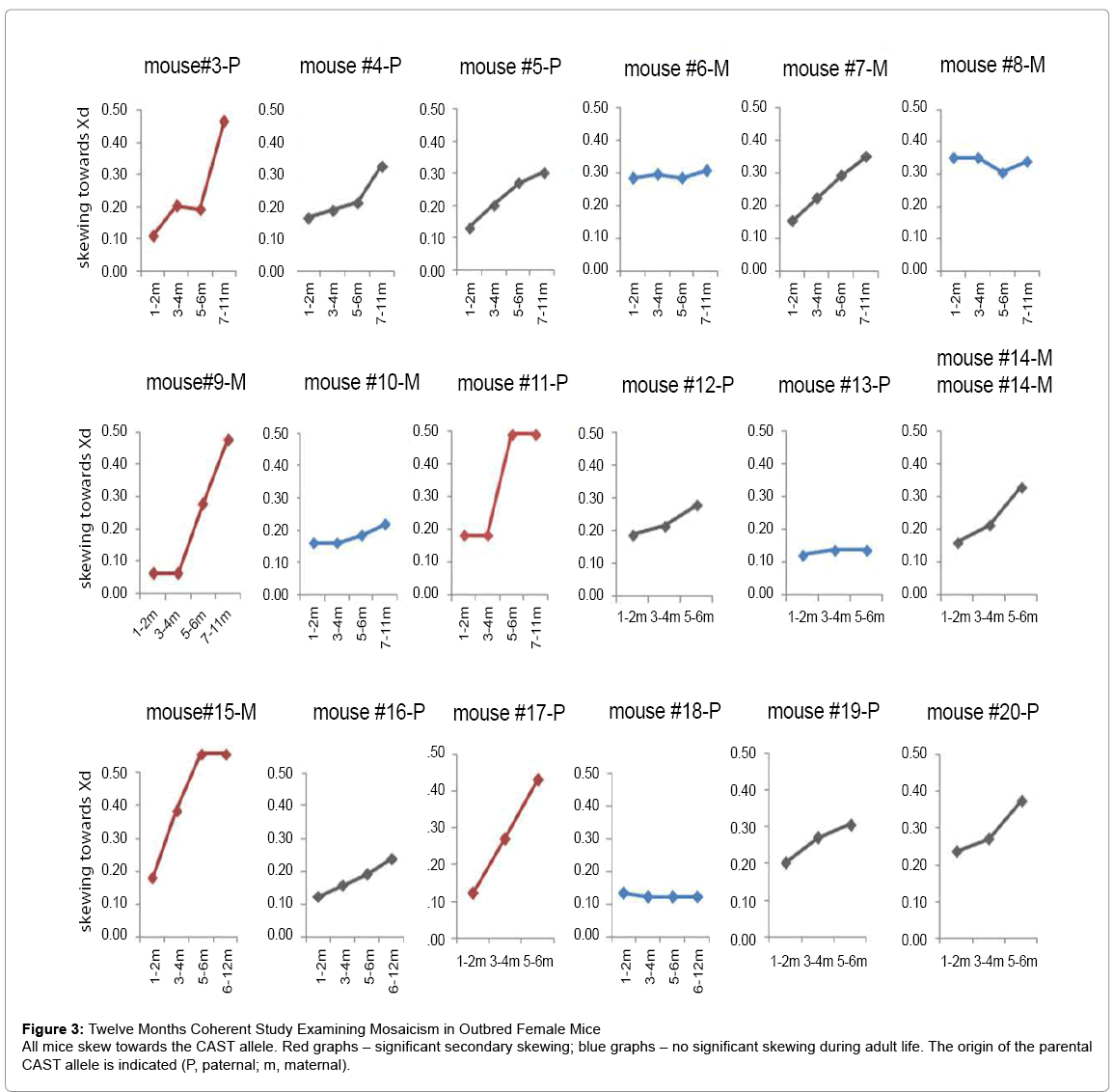
Figure 3: Twelve Months Coherent Study Examining Mosaicism in Outbred Female Mice
All mice skew towards the CAST allele. Red graphs – significant secondary skewing; blue graphs – no significant skewing during adult life. The origin of the parental CAST allele is indicated (P, paternal; m, maternal).
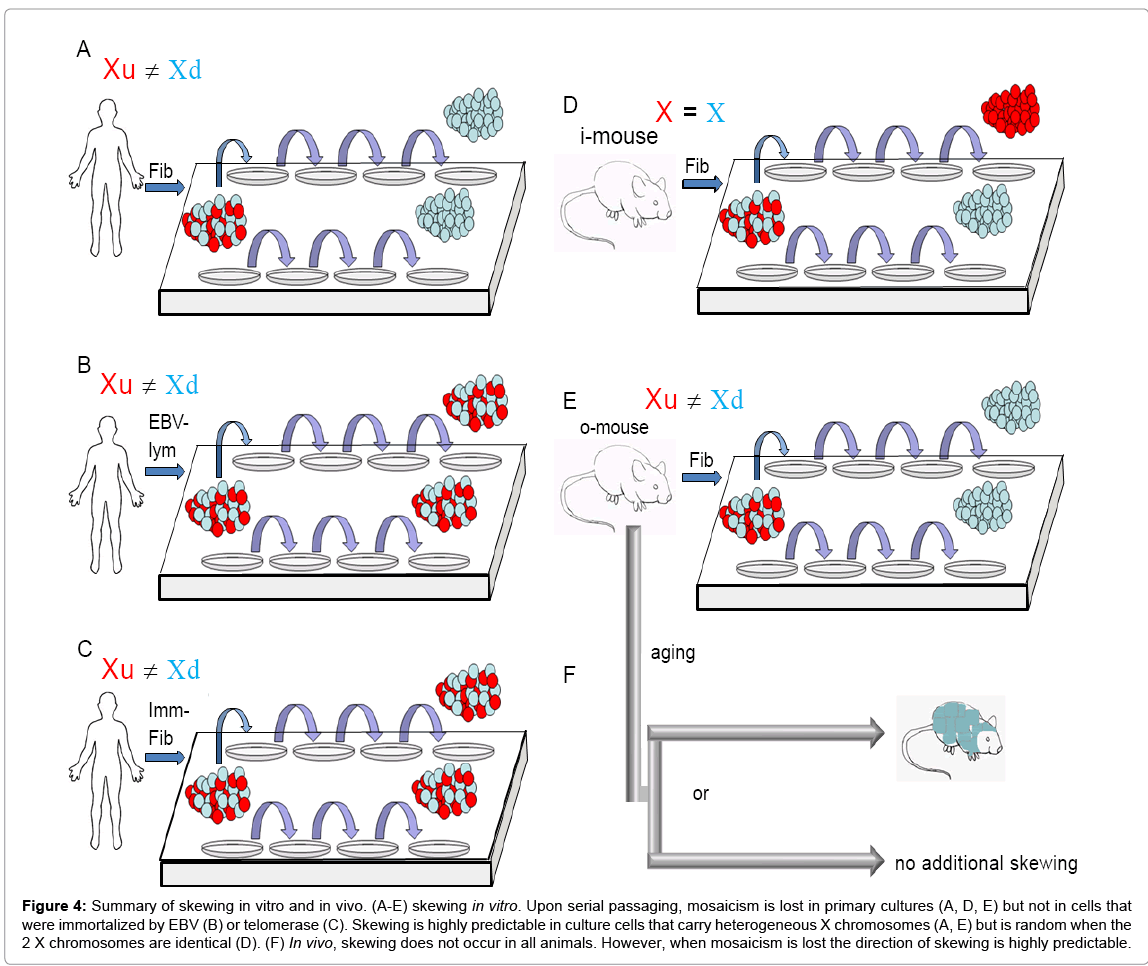
Figure 4: Summary of skewing in vitro and in vivo. (A-E) skewing in vitro. Upon serial passaging, mosaicism is lost in primary cultures (A, D, E) but not in cells that were immortalized by EBV (B) or telomerase (C). Skewing is highly predictable in culture cells that carry heterogeneous X chromosomes (A, E) but is random when the 2 X chromosomes are identical (D). (F) In vivo, skewing does not occur in all animals. However, when mosaicism is lost the direction of skewing is highly predictable.
Implications of skewing in vitro
Primary HFF have been widely used for long-term culture studies in cancer and senescence (e.g. WI-38 and IMR-90). Our finding that the proportion of cells expressing a particular X chromosome gradually shifts over the course of a study introduces a hitherto unconsidered variable into any experimental interpretation and points to the more prudent choice of male cells for future analyses. On the other hand, we show here that mosaicism is relatively stable in EBV-transformed lymphoblastoid cell lines, suggesting that in contrast to primary cells, skewing is less of a concern when using established cell lines.
In some studies, the mosaic nature of females may be an advantage. For example, when modeling X-linked diseases using iPSCs, the coexistence of cells exclusively expressing the wild type or the mutant gene allows the generation of pure populations of mutant iPSCs and isogenic wildtype controls from the same patient (e.g. Rett syndrome patients (31). We have previously demonstrated that skewing may pose a barrier to reprogram one of the isogenic populations. Thus, loss of one population has implications for in vitro stem cell field (32).
Skewing has been shown to occur in several X-linked diseases (2,16,17) and ~10% of healthy females, mainly in tissues with high proliferation, such the hematopoietic lineage (14,33). However, to the best of our knowledge, loss of mosaicism in normal dermal fibroblasts has never been reported in vivo, suggesting that HFF usually do not skew under physiological conditions. In agreement, at early passage, all HFF lines used in this study were a balanced mosaic of cells. We speculate that skewing of fibroblasts in vivo is normally prevented by a combination of low proliferation rates and suppressive niche factors. Identifying better culture systems which imitate physiological conditions may potentially alleviate skewing of primary HFF in vitro, or reversing this argument, culture conditions that alleviate skewing are potentially more physiological-like.
Mechanism behind skewing in vitro
Most of the X-linked genes are not involved in sex determination but in various other processes some of which can influence the cell’s proliferation potential. For example DKC1, a telomerase subunit is mutated in dyskeratosis congenital [34] and FANC B encodes a component of a complex involved in the DNA damage response and if mutated results in Fanconi’s anemia [35]. Thus, differences in function and/or expression levels of X-linked associated gene may affect the cells replicative lifespan. In contrast to inbred mice, humans as a species are so outbred that it is estimated that at least 20% of our (~1200) genes on the two X chromosomes differ significantly [36]. Some of these SNPs may affect gene/snRNA regulation as a consequence of polymorphisms in promoter/enhancer sequences, whereas other SNPs may affect transcript splicing or protein function as a result of polymorphism within coding regions. Thus, polymorphisms in some X-linked genes may create proliferative differences between the 2 isogenic populations leading to (nonrandom) skewing with time. Our data argues against other potential explanations such as the involvement of imprinted genes or an elite subpopulation. Unfortunately, since polymorphism may affect multiple genes and at several levels of regulation - transcription, splicing, stability, translation, and function, testing this hypothesis directly by knock-down/over -expression experiments is not feasible. Nevertheless, this theory is in line with in vivo studies that show high correlation in skewing between elderly monozygotic twin pairs [7,9,10] and in the direction of skewing across tissues obtained from the same individual [11-15].
Skewing in vivo
Skewing in vivo can either result through nonrandom choice of X chromosome inactivation during early developmental stages (i.e. primary skewing) or through secondary selection in favor of cells expressing a particular X chromosome. In the mouse, the Xce locus influences the choice of which X will undergo inactivation during early embryonic stages. While in Xce homozygotes, there is an equal probability that either parental X alleles will be inactivated, in Xce heterozygotes one parental X is preferentially inactivated [29]. Since CAST mice were reported to express a stronger Xce than B6 mice [26], offspring of CAST and B6 crossings are expected to preferably inactivate the B6 X allele. Here we show for the first time in mice that in addition to nonrandom X inactivation, skewing can continue during adult life and when it does, its pattern is strikingly predictable.
Both skewing events share the same Xd. However, since it is highly unlikely that the Xce element is involved in age-related skewing, this coincidence probably reflects some growth advantage conferred by other genes on the X chromosomes of the same strain. In agreement, we saw several fundamental differences between primary and secondary skewing. While all animals showed primary skewing with low variability between individuals, not all syngeneic animals skewed during adult life. Furthermore, we did not see any correlation between the degree of primary skewing and the extent of skewing (if any) with age. The factors that are involved in secondary skewing remain unidentified. Our findings that secondary skewing in mice is strain, but not parental related suggest that skewing of the hematopoietic system is not stochastic and that in contrast to other tissues [24,25] X-linked imprinted genes are not involved. We speculate that, similar to the situation in vitro, the mechanisms behind the predicted direction during age -related skewing in vivo is related to X-linked polymorphism. Additional studies are required to confirm this hypothesis. In humans, there is a strong correlation between skewing and the probability of developing several devastating diseases [2,16-18,21,20]. This correlation may result by a “shared cause”, such as chromosomal anomalies which lead to both skewing and cancer. In agreement, many of the anomalies seen in elderly skewed females are characteristic of those found in hematological cancers and identify common deleted regions within genes previously associated with these cancers [21]. Alternatively, the basis for the correlation between skewing and some diseases may be a “cause and a consequence” as was suggested by Stewart [37]. Identifying environmental and/or stochastic factors that lead to age-related skewing may give us new insights into several life threatening conditions. Our findings that skewing in adult outbred mice occurs in a nonrandom pattern towards a dominant population are in agreement with a previous study in outbred cats [38] and together imply that this may be the situation in humans as well. Our findings suggest that mice, which have relative short life span and may undergo skewing within less than 12 months, may serve as an animal model for this line of study.
Human and mouse Fibroblasts
C57BL/6 carrying GFP on one X allele (B6-GFP) were generated by backcrossing Tg(ACTB-EGFP)D4Nagy/J mice with C57BL/6 for 5 generations. Outbred mice were generated by crossing B6-GFP with CAST/EiJ. Fibroblasts were extracted from E12.5 fetuses. Inbred mice were a generous gift from Dr. Adrian Bird, University of Edinburgh. All fibroblasts were maintained in DMEM supplemented with 15% fetal bovine serum, 2 mM L-glutamine, 1% NEAA, and antibiotic (10 mg/ ml penicillin and streptomycin). For a list of all fibroblasts see Table S1.
6TG and HAT selections
To produce isogenic populations from HPRT+/- Lesch-Nyhan carrier (Coriell: GM01661), cells were incubated with media containing either 6TG (30–60 μM 2-amino-6-mercaptopurine; Sigma) or HAT (1×10-4 M hypoxanthine, 4×10-7 M aminopterin, 1.6×10-5 M thymidine; Invitrogen) respectively, for a period of 6 days and then incubated in normal fibroblast medium.
RNA Isolation, Reverse Transcription, and SNP Analysis
Total RNA was extracted using the Nucleospin RNAII Kit (Macherey-Nagel) with DNase digestion. RNA was quantified using ND-1000 spectrophotometer (Biofrontier Technology) and first strand cDNA was produced with M-MulV reverse transcriptase (Biolabs) using 1 mg of total RNA input. PCR was performed using Supermix system (Invitrogen) . Primer sequences are listed in Table S1. To identify SNPs, PCR product were either sent to 1ST Base for sequencing analysis or analyzed by restriction digest as indicated in Table S1.
Immunostaining
Cells were fixed in 4% paraformaldehyde in PBS at room temperature and blocked in 4% fetal calf serum with 0.1% Tween 20 for 60 min at room temperature. Cells were then stained with primary and secondary antibodies (Invitrogen and Alexa Fluor, respectively) according to standard protocols. Primary antibodies used were as follows: GFP (Abcam, ab290), Lamp2 (Abcam, ab25631). Images were captured with a Zeiss axiovert 200 microscope. Images were enhanced using Paint Shop Pro software and processed evenly across the entire field using Paint.
We thank Adrian Bird for providing primary mouse cells, Barbara Knowles for providing mice, Ray Dunn and Bruno Reversade for helpful advice.
All experiments were performed by Oz Pomp and Denise Fong Mei Leong. Pamela Ying-Yuan Mok performed dynamic assays for SF9 cells. Jerry Chan provided human fetus material. The work was performed under the supervision of Alan Colman.