Rheumatology: Current Research
Open Access
ISSN: 2161-1149 (Printed)
ISSN: 2161-1149 (Printed)
Research Article - (2012) Volume 0, Issue 0
Objectives: The application of mesenchymal stromal precursor cells (MSC) in clinical settings requires a pharmaceutical definition of cell quality including identity, purity, and potency of the individual preparations. In vitro chondrogenesis is presented to define identity and potency independently of specific laboratory conditions.
Methods: MSC obtained from 34 patients undergoing hip replacement surgeries were expanded and brought to chondrogenic differentiation in two laboratories in the USA and in Germany. Cell culture protocols were modeled along established procedures with local variations (primary isolation, serum, hormones, micromass and hydrogel cultures). Chondrogenic differentiation in micromasses and in hydrogel encapsulated cells was analyzed by mRNA quantification, and by determination of glycosaminoglycan content.
Results and Conclusions: The tested cell populations did not differ significantly in the patterns of differentiation between the two laboratories, indicating a strong genetic stability of the adaptive properties of the tested MSC. The results suggest that chondrogenesis may serve as a performance predictor in measures towards standardized quality control.
<Keywords: Mesenchymal stromal cells; Chondrogenesis; Quality control; Hydrogel
Mesenchymal stromal precursor cells (MSCs) represent a promising candidate cell population as a component of Tissue Engineered Medicinal Products (TEMPs, U.S.A.) or Advanced Therapy Medicinal Products (ATMPs, E.U.) [1-3]. In vitro isolation, propagation, and differentiation protocols are the core technologies employed to exploit the functional properties of MSCs. These protocols serve specific purposes, such as optimizing conditions for cell expansion to facilitate subsequent transition of the MSCs to bone, cartilage, or fat cells. While the original protocols, such as the chondrogenic differentiation protocol by Johnstone et al. [4], are still practiced, many laboratories have since developed individually modified recipes. Not withstanding these variations, much of the scientific discussion on the functional characteristics and properties of MSCs has continued with little consideration of the different experimental and technical backgrounds. It is thus likely that current controversies related to data sets and other issues may have been influenced in part by variations in the technical and experimental circumstances rather than solely a consequence of the biologic variabilities of MSCs.
Point in case is donor age-dependent proliferation and differentiation capacity. Published data and their interpretation range from implying a full dependence on age of all MSC properties via demonstrating the age dependent frequency of MSC with otherwise uncompromised biological potential to observing that the population of MSC in a given tissue compartment (mostly bone marrow) is ageindependent [5-11]. Typically, those contributions do not compare different tissue niches (iliac crest vs. humerus vs. adipose tissue) within one donor. Although a theoretical scientific discussion takes place, the status of those considerations remains opinion-based. However, elements such as tissue origin may have considerable influence on the observations and are crucial towards standardization in a regulated environment.
Currently, a heterogeneous collection of cells, derived from a variety of tissues from the body, is the starting cell population by the majority of scientists. While this procedure may be sufficient to allow for the analysis of general MSC properties, such a set-up does not comply with the rules set forth by the authorities: identity, purity, and potency to be defined with respect to each particular application and individual patient. In other words, a random assortment of cells will not be considered as permissible because it implies that a great number of cells given to the patient will not contribute to the intended cure but, in the best case, simply perish, and in worst case, generate undesirable side effects. The current situation is comprehensively described within a review article by Wagner and Ho [12]. The author concludes that “The recent controversy on the multiline age differentiation potentials of some specific MSC preparations might be attributed to this lack of common standards. More precise molecular and cellular markers to define subsets of MSC and to standardize the protocols for expansion of MSC are urgently needed” [12]. These scientific findings on the properties of adult MSCs are being critically evaluated in the application of MSCs for regenerative therapies. Current regulative environment for cell-based therapies assumes a strict consent situation concerning the quality assurance measures for cells, similar to those that exist for classical drugs [1-3]. However, in contrast to classical drugs, where parameters such as molecular weight, metabolism etc. are defined and secured by internationally validated procedures and standards, cell biology to-date does not provide for such calibrated and by-consent-validated systems as TEMPs are, and is still largely viewed as a non-regulated basic science environment.
To fulfill the requirements of regulating authorities, measures have to be implemented that soundly indicate collected cell populations perform as intended. In the absence of yet-to-come perfect screening and sorting procedures, another argument has to be brought forward to support the validity and safety of a given treatment: the assessment of failure risk.
For this reason, comparative research approaches are critically needed that clearly define such parameters. However, variations in protocols used in different laboratories have complicated clear definition of baseline properties of MSCs since the possible ranges in cell quality originating from those protocols cannot be accurately assessed. In this report, we present our attempt to estimate the potential range of quality of MSCs, based on chondrogenic differentiation as a model activity, using cells and methods established in two independent laboratory environments in Germany and in the U.S.A.
A collaborative research undertaking enabled the two laboratories to compare baseline data on the establishment and propagation of human MSCs, and to compare their methods to induce chondrogenic differentiation evaluated on the basis of mRNA expression profiles of a number of chondrogenesis-associated genes and the production of cartilage extracellular matrix (ECM). Modifications of the differentiation protocols have been included to assess the importance of specific aspects.
In the present manuscript, two settings are designed to probe the risk of failure, modeled along chondrogenesis. One situation is the change of laboratory, including all the subtle differences associated with such a change: different sera, different providers of reagents, different practical handling (time needed for passaging, etc). Technically standardized, “normalized”, procedures intend to avoid these fluctuations but they have not been assessed within a direct comparison. The other situation is the issue of different technical environments for the cells during homing and differentiation. This situation is challenged by comparing radically different concepts: the micromass culture system with cell-cell contact and interactions [4], and hydrogel systems with no cell-cell contact - the gelatin hydrogel with all the bioactive collagen peptides automatically present, and the albumin hydrogel, containing no bioactive signals [13]. In addition, the signaling factor environment is modulated by applying different growth factors, and introducing hyaluronan to provide selective anchorage for CD44 (an MSC-associated cell surface marker) and secondary support such as scavenging oxygenating radicals.
The data presented here suggest a risk assessment statement is possible: MSC collected in the described way, propagated and then put to work (differentiation) will function within a wide range of environmental settings. Taken together, our findings indicate that the intrinsic chondrogenic differentiation capacity of MSC is a geographic location- and donor-independent parameter to assess the stemness quality of particular MSC preparations.
Isolation of mesenchymal stromal precursor cells (MSCs) from bone marrow aspirates using density gradient centrifugation and expansion with PDGF-BB and EGF (“NMI cells”)
Bone marrow aspirates from patients undergoing total hip replacement were obtained from the BG Trauma Clinic Tübingen. All procedures were approved by the local ethics committee. Informed consent was given for use of the MSCs for research. The mononuclear cell fraction was isolated using Ficoll-paque plus (GE Healthcare Europe GmbH, Freiburg, Germany) density gradient centrifugation for 30 min at 400 g. Isolated cells were seeded into cell culture flasks.
Adherent cells (MSCs) were expanded in expansion medium [9] consisting of 60% low glucose Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Karlsruhe, Germany) 40% MCDB 201 (Sigma- Aldrich, Steinheim, Germany), penicillin/streptomycin (Invitrogen), 0.05 μM dexamethasone, ITS+1 supplement, 0.1 mM L-ascorbic acid 2-phosphate (all from Sigma-Aldrich), 2% fetal bovine serum (Biochrom AG, Berlin, Germany), 10 ng/ml platelet-derived growth factor BB (PDGF-BB), and 10 ng/ml epidermal growth factor (EGF) (both from Miltenyi Biotech, Bergisch Gladbach, Germany). When MSCs reached approximately 80% confluence, they were detached with 0.25% trypsin/EDTA (PAA Laboratories GmbH, Pasching, Germany) and were either used for gene expression analysis, differentiation experiment or stored in liquid nitrogen. If thawed cells were used for chondrogenic differentiation experiments they were expanded for one additional passage. For differentiation cells were mostly used in passage 2, in some cases passage 3 was used. Totally 23 donors are included in the experiments (10 females, mean age: 74.3 years; 13 males, mean age: 53.2 years).
Isolation of mesenchymal stromal precursor cells (MSCs) from trabecular bone marrow and expansion with FGF-2 (“Pittsburgh cells”)
Trabecular bone pieces were harvested from donor femurs using a bone tong. Bone pieces were transferred in a Petri dish and minced with a scissors. The bone marrow released during this procedure was sieved through a 40 μm cell strainer. For further collection of bone marrow, bone pieces were washed with α-MEM (Minimal Essential Medium) and the washing medium was also sieved through a cell strainer. The suspension was centrifuged at 300 x g, 5 min. and the obtained cell pellet was washed twice with α-MEM. Cell numbers were determined and the cells were cultured in α-MEM low glucose (Invitrogen/Gibco), 10% FBS (MSC qualified, Invitrogen/Gibco), 1% Antibiotic-Antimycotic (Invitrogen/Gibco), and 1 ng/ml basic fibroblast growth factor (FGF- 2) (R&D Systems). In total, cells from 11 donors were included in the experiments (4 females, mean age: 60.5 years; 7 males, mean age: 56.9 years), and from passage 2-3.
Induction of chondrogenic differentiation
MSCs were differentiated chondrogenically as micromasses (MM) and in different types of hydrogels, applying a standard induction protocol [4].
Micromasses were produced by pipetting 20 μl of the MSC suspension, containing 0.4 x 106 cells, into an uncoated well of a 96-well plate [4,14]. Medium was added to the cells after two hours. Three days later the generated MM was transferred into a well of a 12-well plate.
In order to achieve chondrogenic differentiation in hydrogels, 0.4 x 106 MSCs were embedded in 500 μl gelatin-based hydrogel or human serum albumin- based matrices. Gelatin hydrogels containing 10% gelatin from pig skin (provided by Gelita AG, Eberbach, Germany) and gelatin-hyaluronan hydrogel containing 10% gelatin and 3.5 mg/ml hyaluronan (HA; Visiol, 20 mg/ml, TRB Chemedica AG, Haar, Germany) were used. The gelatin was enzymatically crosslinked by adding 0.15 U transglutaminase. Polymerization time was approximately 30 min.
Human serum albumin (HSA) and HSA-HA were produced as described elsewhere [13]. The HSA hydrogel used here was based on a chemically activated human maleolyl-albumin which was cross-linked by a specific thio-polyethylene glycol (SH-PEG) (pending patents no. PCT/EP2008/005643, DPMA 10 2008 008 071.3).
Briefly, to prepare 1ml cell-containing HSA hydrogel, trypsinated cells were counted and a pellet with 0.8 x 106 cells was made by centrifugation. The cell pellet was resuspended in 730 μl culture medium and 70 μl chemically activated maleolyl-albumin (HSA) stock solution were added to the cell suspension. The proportion of cellcontaining HSA to stabilized SH-PEG cross-linker stock solution was 4:1 (v/v).
To prepare 1 ml cell-containing HSA-HA hydrogel, 70 μl chemically activated maleolyl-albumin (HSA) stock solution, 200 μl high molecular weight hyaluronan (HA; Visiol, 20 mg/ml, TRB Chemedica AG, Haar, Germany) and 400 μl culture medium were mixed and incubated for 1 h at 4°C.
The cell pellet was resuspended in 130 μl culture medium and then added to the HSA-HA mixture. The proportion of cell-containing HSA-HA to stabilized SH-PEG cross-linker stock solution was 4:1 (v/v). The cell-containing gel mixture was filled in the larger chamber of a two-chamber syringe, the cross-linker in the smaller one. This dual syringe consists of two chambers holding a dual linked piston with a separate outlet for each chamber. The chambers are to be filled reversely via the outlet. After filling, the outlets are closed with a snapin mixing chamber. The chamber combines the outlets and guides the components within the two chambers together into a common tube. The tube is equipped with an interrupted 12 turn-helix that intertwirls the content of the two chambers. The dimensions of this mixer are designed to match the viscosities of the polyethylene glycol and the HSA-HA.
Hydrogels with 500 μl containing 4 x 105 MSCs were produced by injecting the content of the syringe via a mixing head and a 18-gauge needle into culture wells. Gelation time was approximately 5 min. After cross-linking, 1.5 ml of the culture medium was added. Hydrogels were cultured at 37°C in humidified atmosphere and 5% CO2.
MSCs were differentiated in chondrogenic differentiation medium [9] consisting of high glucose DMEM, 1 mM sodium pyruvate, penicillin/streptomycin (all from Invitrogen), 0.1 μM dexamethasone, ITS+1 supplement (Insulin-Transferrin-Selenium), 0.17 mM L-ascorbic acid 2-phosphate, 0.35 mM L-proline (all from Sigma-Aldrich), and 10 ng/ml transforming growth factor β3 (TGF- β3, Miltenyi Biotech). The medium was renewed twice a week.
Gene expression in the chondrogenically induced cultures of MSCs was analyzed in for at least 2 weeks (mRNA expression remained stable after 2 weeks); sGAG content was analyzed in MSCs differentiated for four weeks.
mRNA isolation and reverse transcription
Gelatin hydrogels with chondrogenically differentiated MSCs were digested with 4 mg/ml collagenase B (Roche, Penzberg, Germany) at 37°C; HSA hydrogels with 3 mg/ml proteinase K (Sigma-Aldrich). After centrifugation the cell pellet was lyzed in Buffer RLT (Qiagen, Hilden, Germany). MMs were ground with Molecular Grinding Resin from G-Biosciences (Maryland Heights, USA) and directly lyzed in Buffer RLT. MSC monolayers before induction served as a control. After detaching the cell with trypsin/EDTA the cell suspension was centrifuged and the cell pellet was lyzed in Buffer RLT. RNA was isolated with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. An on-column DNase digestion step with the RNase-Free DNase Set (Qiagen) was added during RNA purification. The Reverse Transcriptase Core Kit from Eurogentec (Cologne, Germany) with oligo d(T)15VN primers was used for reverse transcription (RT) according to the manufacturer’s recommendations. A maximum of 1 μg mRNA was transcribed into cDNA.
RT-qPCR
For quantitative PCR (qPCR) it is necessary to have appropriate reference genes that show no expression variations when exposed to different culture conditions. In this paper we examined MSCs cultivated under a variety of culture conditions. They were cultured in monolayers as well as in different three-dimensional cultures, and were differentiated chondrogenically. To determine an appropriate reference gene which is stably expressed under this variety of conditions, we analyzed the expression of five genes (YWHAZ, GAPDH, HPRT I, UBC, and β-actin) biostatistically. The analyses were performed with geNorm software (http://medgen.ugent.be/genorm) [15]. YWHAZ [15] showed the fewest changes in its expression under all tested conditions (data not shown). For this reason we selected YWHAZ for use as the reference gene in this study. Our results were confirmed by Fink and colleagues, who also identified YWHAZ as the appropriate reference gene for the differentiation of human adipose-derived stem cells under hypoxic conditions [16]. The qPCR Master Mix Plus w/o UNG for SYBR Assay Low ROX (Eurogentec, Cologne, Germany) was used for reverse transcription quantitative polymerase chain reaction (RT-qPCR). The PCR was run and analyzed on an Applied Biosystems 7500 Fast Real Time PCR System with 7500 Fast System SDS Software (Applied Biosystems Inc, Foster City, USA). mRNA expression levels were analyzed for collagen type I (COL1A2), collagen type II (COL2A1), aggrecan (ACAN), melanoma inhibitory activity (MIA), collagen type X (COL10A1), and alkaline phosphatase (ALP). Primers were used at 300 nM (Table 1). The specificity of all primers was confirmed via BLAST analysis [17]. PCR efficiencies (E) were calculated for each primer pair using a calibration curve. Quantification cycles (Cq) were determined for each gene. Relative gene expression was calculated according to the following equation: relative mRNA expression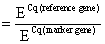 Relative COL2A1 mRNA expressions in MSC monolayers were defined as 10-7 when no amplification signal was detectable.
Relative COL2A1 mRNA expressions in MSC monolayers were defined as 10-7 when no amplification signal was detectable.
| Gene | abbreviation | forward primer | reverse primer | efficiency | Reference sequence |
|---|---|---|---|---|---|
| tyrosine 3-monooxygenase/ tryptophan 5-monooxygenase activation protein, zeta polypeptide | YWHAZ | ACTTTTGGTACATTGTGGCTTCAA | CCGCCAGGACAAACCAGTAT | 1.89 | NM_003406 |
| collagen, type I, alpha 2 | COL1A2 | GCTGGCAGCCAGTTTGAATATAAT | CAGGCGCATGAAGGCAAGT | 1.96 | NM_000089 |
| collagen, type II, alpha 1 | COL2A1 | AGAGGTATAATGATAAGGATGTGTGGAAG | GTCGTCGCAGAGGACAGTCC | 1.94 | NM_001844 |
| aggrecan | ACAN | TGCATTCCACGAAGCTAACCTT | GACGCCTCGCCTTCTTGAA | 1.98 | NM_013227 |
| melanoma inhibitory activity | MIA | CCCAGTAGCATTGTCCGAGA | GGCAGTAGAAATCCCATTTGTCT | 1.91 | NM_006533 |
| alkaline phosphatase | ALP | TTCCCACGTCTTCACATTTGG | TTGCCATACAGGATGGCAGTG | 1.95 | NM_000478 |
| collagen, type X, alpha 1 | COL10A1 | CACGCAGAATCCATCTGAGAAT | CGTTCAGCGTAAAACACTCCAT | 1.96 | NM_000493 |
Table 1: Sequences and efficiencies of primers for RT-qPCR analysis.
Sulfated glycosaminoglycan (sGAG) quantification
To determine the amount sGAG released into the culture supernatant during 4 weeks of differentiation a Blyscan Glycosaminoglycan assay (Biocolor Life Science Assays, Carrick, UK) was performed according to the manufacturer’s instructions. Chondroitin sulfate provided in the kit was used as standard.
Statistical analyses
Statistical analyses were performed with Sigma Stat 3.0. P-values were determined with t-test when two normally distributed groups with equal variances were compared; if not rank sum test was used (Figure 1 and Figure 2). For multiple comparisons in Figure 3 One Way Analysis on Variance (ANOVA) was performed. To obtain normally distributed data, the data set was logarithmized. If data did not pass the equal variance test Kruskal-Wallis ANOVA on Ranks with subsequent multiple comparison procedure (Dunn´s method) versus control group was performed. P-values smaller than 0.05 were considered as statistically significant and were marked with an asterisk.

Figure 1: Display of baseline gene expression (A) and size (B) of the Pitt (light gray boxes) and NMI (dark gray boxes) cells during propagation in vitro. There is a certain tendency for the Pittsburgh cells towards early chondrogenic differentiation but the levels are still 2 to 3 orders of magnitude lower than after induction of differentiation (see Figure 2). (* = significant, p<0.05, t-test if normality and equal variance tests are passed, if not, rank sum test was performed.) Bars = 500 μm.

Figure 2: Cell properties after induction of differentiation. A: gene expression profile of the Pitt (light gray boxes) and NMI (dark gray boxes) cells. B: typical size difference of the differentiated micromasses (left: Pittsburgh cells, right: NMI cells). C: sGAG content in culture supernatant for two donors (Pitt, light gray boxes; NMI, dark gray boxes). Both Pitt and NMI cell preparations differentiate equally well, there is a slight tendency for the NMI cells towards higher Col2 expression (* =significant, p<0.05, t-test if normality and equal variance tests are passed, if not rank sum test was performed). Bars = 500 μm.

Figure 3: A. Modification of gene expression patterns under different culture condition (MSC – undifferentiated monolayer, MM – differentiated micromass culture, Gel – gelatin hydrogel, GHA – gelatin hydrogel with high molecular weight hyaluronan incorporated, HSA – human serum albumin hydrogel, HSA-HA – human serum albumin hydrogel with high molecular weight albumin incorporated. After four weeks of chondrogenic differentiation there is generally no difference in the gene expression levels among the five three-dimensional cultures, with the exception of collagen type 1 expression in gelatin hydrogels (* = significant, p<0.05, one way ANOVA on ranks). B. Morphological appearance of MSCs chondrogenically differentiated in Gel (top) and HSA (bottom) hydrogels. Bars = 500 μm.
MSC phenotype is influenced by different isolation and expansion protocols
To investigate the influence of different isolation and expansion protocols on the gene expression of the cell cultures obtained from these different protocols, we compared the morphological appearance of the cells and the expression of six different genes. Features of both protocols are summarized in Table 2.
| Cell source | Cell status at harvest |
Purification/ enrichment |
Plating | Expansion | Appearance after expansion | |
|---|---|---|---|---|---|---|
| NMI protocol | bone marrow aspirate from femur shaft | in suspension | density gradient centrifugation | mononuclear cell fraction | 60 % DMEM low glucose, 40 % MCDB-201, 2 % FCS, 1x ITS+1, 1.1 mM ascorbate, 0.05 µM dexamethasone, 10 ng/ml PDGF-BB, 10 ng/ml EGF [ Reyes 2001] |
heterogeneous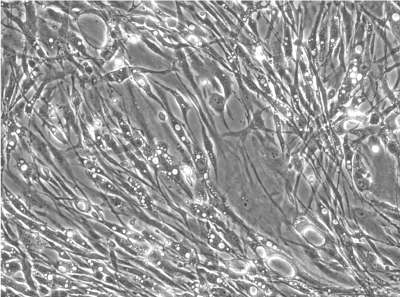 |
| Pittsburgh protocol | trabecular bone marrow from femoral head | adherent | none | whole cell fraction | a-MEM, 10 % FBS (MSC qualified), 1 ng/ml FGF-2 |
homogeneous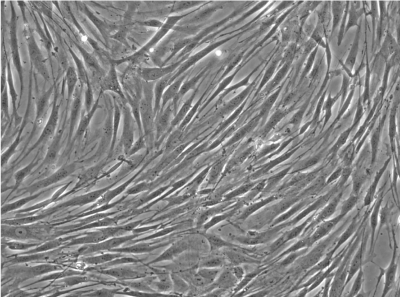 |
Table 2: Two isolation and expansion protocols of human bone marrow MSCs.
The chondrogenic markers collagen type 2 (COL2A1), aggrecan (ACAN), the transcription factor melanoma inhibitory activity (MIA, also known as cartilage-derived retinoic acid-sensitive protein CDRAP), and collagen type 1 (COL1A2), collagen type 10 (COL10), and alkaline phosphatase (ALP), representing more osteogenic markers, were analyzed and their expression calculated relative to the reference gene YWHAZ (tyrosine 3-monooxygenase) (Figure 1).
Cells isolated and expanded according the “NMI protocol” (NMI cells) have been characterized by Scharstuhl et al. [9]. The cells showed the typical surface antigen phenotype for MSC, being positive for CD10, CD 73, CD 90, CD 105, CD 140b, and CD 166, and negative for CD 3, CD 14, CD 16, CD 20, CD34, CD45, CD 235. Cells isolated and expanded according the Pittsburgh protocol (Pittsburgh cells, laboratory of Dr. Rocky Tuan). These cells displayed MSC capabilities, characterized by the ability to proliferate extensively while remaining the potential to differentiate along the osteoblastic, adipogenic, and chondrogenic lineages. The trabecular bone marrow derived cells were positive for the surface antigens CD44, CD73, CD 105, STRO- 1, CD146 and negative for CD14, CD19, CD 34, CD 45, CD79a and CD144. Besides a similar surface antigen profile, the cells differed from each other in their morphological appearance. In the cell suspension directly after detaching the cells from the culture plastic, the Pittsburgh cells were homogenous in form and shape, whereas the NMI cells appeared very heterogeneous (Figure 1B). Significant differences were also found in the gene expression profiles of both cell types (Figure 1A). Pittsburgh cells showed significantly higher expression levels for ACAN (1 x 10-3 vs. 5 x 10-5), MIA (2 x 10-2 vs. 3 x 10-4), and COLX (1 x 10-3 vs. 2 x 10-5) compared to the NMI cells. In contrast the expression of alkaline phosphatase was significantly lower in Pittsburgh cells (2 x 10-2 vs. 4 x 10-1). No differences were observed in the expression of COL2A1 and COL1A2.
Similar chondrogenic response to induction by transforming growth factor β3 in initially different expanded cells
Cells obtained from the different isolation and expansion protocols were then examined regarding the chondrogenic differentiation potential in micromass cultures. Chondrogenic induction was induced by transforming growth factor β3 (TGF-β3). Here too a difference between the two different cell preparations was seen in the morphological appearance/size of the micromasses (Figure 2B). The Pittsburgh cells formed smaller and more compact micromasses, whereas the NMI cell micromasses were larger but less condensed. Chondrogenic induction was successful in both cell preparations, as evidenced by significantly increased expression of the chondrogenic marker genes COL2A1, ACAN, and MIA, but also the hypertrophic cartilage marker COL10. Interestingly, after two weeks of chondrogenic differentiation the gene expression profile was much more homogeneous than after expansion (Figure 2A). Only COL2A1 was significantly different between micromasses derived from Pittsburgh cells and the micromasses obtained from NMI cells: NMI: 2 x 10-3; Pitt: 5 x 10-4. From two donors it was possible to isolate and expand the cells in parallel according to both the Pitt and the NMI protocol. These cells were chondrogenically differentiated under standard conditions in micromasses and the sGAG released in the culture medium was examined (Figure 2C). In both cases, the Pittsburgh cells released slightly more sGAG into the culture supernatant than the NMI cells did.
Different three-dimensional environment allow chondrogenic differentiation with similar outcome in gene expression profile
For the application of MSCs in regenerative cell therapies a convenient system for cell delivery and cell anchorage must be provided. Hydrogels are considered promising tools for this purpose. Therefore four different hydrogels were used for chondrogenic differentiation. In order to examine whether the three-dimensional environment of different hydrogels is also able to affect the differentiation process, or whether TGF-β3 alone is responsible for the outcome of the differentiation, chondrogenic differentiation in four different hydrogels was compared with differentiation in the classical micromass model (MM). In contrast to micromasses, in which cells are densely packed, in hydrogels the cells are embedded with almost no direct cell-cell contacts [18]. The chosen hydrogels are based on two different materials, possessing totally different properties in terms of their interaction with cells. One base material is gelatin (“Gel”, from pig skin), which allows cell adhesion. The other base material is HSA, which does not mediate any specific cell adhesion. Both materials were used to prepare hydrogels with and without high molecular weight HA (“GHA”, or “HSA-HA”, respectively). The different appearance of the cells in hydrogels made from gelatin and from human serum albumin can be seen in Figure 3B. In gelatin hydrogels the cells appeared spread and attached to the matrix, whereas in the HSA hydrogels the cells displayed a round shape without any cellular outgrowths.
In all hydrogel types chondrogenesis was successfully induced with TGF-β3 and the gene expression profiles were comparable to those in micromasses. As a result of TFGβ induction, a significant increase in chondrocyte specific genes COL2A1, ACAN, MIA was found. In addition, the hypertrophy marker COL10 mRNA was raised in all hydrogels and micromasses compared to the MSC prior to induction. The stimulation of these genes was between about 1,000 to 10,000-fold for all 3D cultures compared to the initial MSC monolayers. Strongest induction was seen for COL2A1 with a mean increase in mRNA transcription of 9,700-fold (mean of all 3D cultures compared to mean of MSC). Mathematically significant differences were also found for COL1A2 expression in 4 out of 5 of the 3-dimensional culture systems compared to MSC (MM, Gel, Gel-HA, and HSA-HA). As the mean difference was only about 5-fold, this might not be biologically relevant. The mRNA transcription rate of ALP was unchanged upon the induction of chondrogenesis by TGF-β3 and unaffected by the three-dimensional environment. Of the five different gel cultures systems, only one significant difference was found. The expression of COL1A2 in gelatin hydrogels (Gel, mean: 8.05 x 101) was slightly higher than in micromasses (MM, mean: 3.4 x 101), but this 2.4-fold increase was unlikely to have biological relevance.
With the first attempts to introduce MSCs in regenerative treatment patterns, perhaps most spectacularly in heart diseases [19,20], the issue of uniform standardization has gained more and more attention. Oversight agencies have drafted legal frameworks and definitions (TEMP = tissue engineered medicinal products (USA), ATMP = advanced therapies medicinal products (EU)) but their conversion into measurable methodology still remains to be implemented. Advisory bodies such as ASTM in the USA or DIN/ISO (Deutsche Industrienorm/ International Standard Organization) committees in the EU and elsewhere are struggling to find cornerstones for quality assurance and application safety.
One particular problem is the level of reproducibility. This is fact initially viewed somewhat of a surprise in the field of cell biology, since reproducibility as a hallmark of good science practice often does not necessarily include quantitative aspects. Cell biology, derived from cytology and histology, is very much committed to the exploration of biological principles, with the quantitative aspects of cellular reactions often treated secondary versus the quality of a cellular response.
However, in regenerative medicine, the amount of healing power is as critical as the right choice of the medicine, i.e. the cell and its supplementary, or galenic, property. There are only few documents publicly available that provide an insight into how for example companies deal with their emerging regenerative therapies towards establishing credibility in terms of quantitative descriptions or their products. This situation leaves the committees and sub-committees of the various guidance bodies struggling with the task to define at least boundary guidelines for the description of quantitative cell properties, including the uncertainty concerning the methods of choice.
The data set presented here sheds some light on the stability of MSC populations, illustrated by their potential to differentiate into cartilaginous cell derivatives. In general, the data reveal rather robust quantitative characteristics when challenged with the typical modifications occurring in different laboratory settings.
The central insight culminates in the proof of principle that MSC lines express stable chondrogenic features in terms of inducibility and extent of chondrogenic differentiation independently of particular laboratory settings. While evidence for such biologic stability was present and could be extracted from literature comparisons, no interlaboratory investigation has attempted to directly test this situation. For the application of a defined procedure under regulated conditions, however, it is imperative that the baseline properties of MSC with regard to biological potential have to be verifiable independent of highly specific milieus. Currently, no standards for quality criteria towards functionality have been generated. The present data set suggest that global standardization of the potential of MSC is possible at least for chondrogenesis. Interestingly, a recent effort has been made towards a proposal for ASTM standardization of an automated colony forming unit assay (WK33534, approved by subcommittee F04.43, George Muschler, Orthopaedic Research Center, Cleveland Clinic, Ohio 44195, USA). The assay is, among others, meant to define the proliferative capacity of MSCs under standard culture conditions.
Concerning donor-to-donor variability, the stable results for the analysis of mRNA transcripts is particular remarkable due to the fact that the cells were not matched, but obtained from quite different donors. Despite well known high donor-to-donor variation, chondrogenic induction with TGF-β3 appeared to be a stimulus strong enough to equalize the gene expression of initially different cells, irrespective of their genetic background, donor age, and in vitro differences with respect to passage number.
Besides this global uniformity of chondrogenic response, some variability was observed that may be related to the laboratory conditions. In one case, a delayed onset of chondrogenesis was observed for NMI cells, from day 3 after induction to day 7, diminishing by day 14 (data not shown). It is therefore suggested that in vitro test periods be 14 days or longer.
Another difference emerged for the expression levels of collagens type I and type II mRNAs after induction (Figure 2 and Figure 3). However, although mathematically significant, the low extent of differences suggests that these are biologically insignificant. Detailed analyses of collagen types 1 and 2 protein content within the micromass cultures would be needed to verify this, as immunohistochemistry is inherently qualitative (sic!).
Besides these changes, almost no differences between micromasses and the different hydrogels are found for any of the analyzed genes. This insensitivity to the environment in which differentiation takes place suggests that the stimulatory signals of TGF-β3 are more important than the type of matrix constituting the three-dimensional environment: cell-cell interaction, collagenous peptides (gelatin), or hyaluronan. This result is particular noteworthy given the quite different cell morphology in the different hydrogels, with predominantly spherical in albumin gels, polygonal in micromasses, and elongated in gelatin. This finding could be of significance in risk assessment for the evaluation of untoward effects induced by such MSC populations with different morphologies.
Many recent studies have proposed additional cell surface markers to identify particular subsets of stromal cells. The goal is to differentiate mesenchymal from epithelial or hematogenic lineages on the basis of the variety of marker antigens or orphan (antigen unknown) markers [9-11]. In addition, these markers may also be utilized to sort defined cells from crude mixtures obtained after extraction from tissues such as bone marrow by fluorescence activated cell sorting or magnetic bead sorting (the herein utilized cells were initially enriched by density gradients and/or differential adhesion to tissue culture plastic). While the markers carry potential to identify cells equipped with metabolic features that principally enable their lineage-specific differentiation. It should be noted that “identity” may not predict the actual therapeutic efficiency (“potency”). Interestingly, in developmental biology the term “potency” is used as a descriptor for the principle, not for the actually expressed efficiency of a cell population, contrary to how the term is used in pharmacology where it serves as a strictly quantitative element. In other words, a regulatory environment would still demand for a potency assay even if the initially detectable features of the isolated cells predict certain capabilities.
At present, the data set presented here, while insufficient to provide the conclusive demonstration of measures to define potency, demonstrates that MSCs may carry features that are very stable even in slightly varying environments. Further development of such comparative effectiveness will help to define the basis for safe and functional applications of cell therapy.
Even though those approaches are attempts to alleviate the current pressure on cell and tissue engineering to comply with regulatory rules, it is obvious that this new field cannot catch up to the historically grown situation of classical pharmacology within a few decades. Alternative paths should therefore be taken to secure patients relying on the new technologies and an unhindered technical progression of the field at the same time. One path could be not to regulate single elements of such a therapy but a cumulative risk assessment. Risk assessment does not attempt to eliminate single risk elements but weighs overall net gains and losses and provides strategic solutions to minimize chances for treatment failure.
In April 2012, the ISO commission in charge votes towards a new guideline ISO/FDIS 13022 “Medical products containing viable human cells – Application of risk management and requirements for processing practices”. Should this standard be implicated, it may well represent a major leap towards improved legal and regulatory acceptance of novel cell based therapies. Our proposed in vitro assay may represent a typical element towards the philosophy of risk assessment in the field.
The inter-laboratory comparison of MSC potency revealed remarkably stable cell features for both proliferation and differentiation. The data set reported here underscores the reproducibility of quantitative descriptors and their utility as quality control measures for individual patients and across production sites.
We thank Dr. Bernhard Schewe and Dr. Peter de Zwart (BG Trauma Clinic Tübingen, Tübingen, Germany) for providing bone marrow samples.
No conflict of interest.
This study was supported from the German Ministry of Education and Research (BMBF grant 0313755, 315579), by the US Department of Defense W81XWH-10-1-0850 and by the Commonwealth of Pennsylvania, Department of Health [21].