Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2018) Volume 7, Issue 3
The Self Nano Emulsifying Drug Delivery Systems (SNEDDS) have gained considerable interest over the past decade for their use in various applications. They have been shown to increase bioavailability and decrease side effects of highly toxic drugs. Non-Steroidal Anti-inflammatory Drugs (NSAIDS) such as Diclofenac Sodium (DFS) lead to major side effect gastric toxicity. The aim of this study was to formulate, characterize and evaluate DFS entrapped SNEDDS. SNEDDS were prepared by emulsion diffusion evaporation technique. DFS formulated SNEDDS showed the least size of 101 ± 3.5 nm, zeta potential of -18.5 ± 1.6 mv and entrapment efficiency of 90.6 ± 2.1 in FESIF respectively. The initial drug release was rapid in FASIF followed by phosphate buffer and the total drug release was high i.e; 74.18% in FASIF followed by phosphate buffer i.e; 70.14%. The drug released for 3-4 hrs approximately in all the media for 50% of release to occur except in SGF and FASGF in which the total percent release itself was 50% till 30 h. A significant reduction in toxicity of DFS was observed for SNEDDS in gastric mucosa when compared to free DFS with a 10 fold decrease in ulcer index of DFS in acute study and a 6.5 fold decrease in ulcer index of DFS in chronic study.
Keywords: Diclofenac sodium; SNEDDS; Release study; Gastric ulcers
Self emulsifying drug delivery systems (SNEDDS) are one of the promising nanocarriers and are isotropic mixtures of oils/lipids, surfactant, co-surfactant, and drug substance with optional solvents/ cosolvents. The principal characteristic is, its ability to form fine o/w nanoemulsions upon reconstitution in aqueous environment and subsequent dilution in gastric fluids, with gastric motility supplying the necessary agitation. SNEDDS are therefore good candidates for the oral delivery of oil soluble drugs, provided they possess adequate solubility in oils/lipids or oils/lipids-surfactant blends [1]. The use of SNEDDS is one of the most interesting approaches to improve the solubility and oral absorption for poorly water-soluble drugs. Now, much more attention has been focused on SNEDDS due to its excellent efficiency in delivering poorly water-soluble drugs and achieving increase in bioavailability. Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used in clinical practice. Many patients require these drugs on a chronic basis to relieve the pain of arthritis. It is well known that these drugs cause serious undesirable side effects. Damage of the gastric mucosa is the most important and common side effect and includes erosions and ulcers [2,3], as well as complications such as bleeding and rarely perforation. Epidemiologic studies have demonstrated increased morbidity and mortality due to NSAID use. In patients with arthritis these drugs are responsible for 2,600 deaths and 20,000 admissions to the hospital every year. Peptic lesions due to diclofenac sodium develop without pain in 50% to 60% of cases, which delays early diagnosis and thus may lead to complications. diclofenac sodium is a non-steroidal anti-inflammatory, analgesic and antipyretic drug and it has a high solubility above pH5. When administered orally, side effects such as gastric-duodenal ulcers and its short half-life are problems in clinical. Various strategies have been explored to avoid NSAIDs related toxicity, including concomitant administration of gastric protectors [4], use of rectal drug delivery systems [5], or modified release formulations [6]. The development of oral controlled and sustained release offers a potential benefit for NSAIDs. The rationale behind it is allowing the release of the drug at a desired rate, providing sustained levels (fewer doses required) and reducing the contact with gastric mucosa (reduced gastric damage). To this end, SNEDDS have raised increased attention due to their properties and benefits for drug pharmaceutical performance (site specific targeting and controlled release) [7]. As it can be observed, major macroscopic alterations were observed in the stomach. More than 10% of patients who receive diclofenac sodium develop an endoscopically visualized ulcer, an incidence at least 5 to 10 times higher than in patients who are not taking diclofenac sodium [8]. Furthermore, elderly users (60 years) appear to be particularly vulnerable, with an estimated mortality of 10%. Since NSAIDs like diclofenac sodium is one of the most widely used, it was interesting to know how nanotechnology would prove beneficial to these drugs. Recently, NSAID loaded nanoparticles were designed and evaluated, where flurbiprofen nanoparticles with suitable size range are envisaged to concentrate at inflammation sites due to increase fragility of blood vessels at those sites and increased aggregation and prostaglandin synthesis [9]. Although diclofenac sodium is a conventional NSAID, it could be fully utilized without harmful side effects if it is properly formulated. The advantages of SNEDDS over conventional dosage forms have been reported [10]. It minimizes the serious gastric irritant side effects of the conventional NSAID preparations. Diclofenac sodium when formulated in goat fat and Tween 65 mixture to develop self emulsifying tablets to reduce gastric ulcers [11]. The NSAIDS which can cause gastric ulcers include, Piroxicam, which is a poor water soluble drug, is incorporated in self emulsifying lipospheres consisting of a mixture of a homolipid from Capra hircus with Tween 65 as the surfactant. These self emulsifying lipospheres had the best performance in terms of anti-inflammatory effect and possibly could be employed in the formulation of self emulsifying lipospheres for oral administration [12]. Indomethacin [13], ibuprofen and ketoprofen [14] are also formulated in SNEDDS. Therefore the establishment of adequate protection of GI membrane from the effect of diclofenac sodium is of paramount importance. Similarly diclofenac loaded PLGA nanoparticles using DMAB of size 92.4 ± 7.6 nm and zeta potential -11.14 ± 0.5mv are found to be the best alternative to existing oral delivery methods and aid in reducing deleterious side effects common to NASID use [15]. Also a recent finding of suppression of indomethacin induced small intestinal inflammation by orally administered redox nanoparticles is found to be a promising approach as nanotherapeutics [16]. Also as per the recent research reports which states that mucosal damage can be prevented with nanoscale systems such as liposomes, nanoparticles and etc would be a great boon with a high potential for industrial manufacturing [17]. On similar lines a study proved that orally administrated indomethacin loaded redox nanoparticles significantly accumulated in the intestinal mucosa and enhanced uptake of indomethacin and a positive sign of suppression of small intestinal inflammation [18]. Recent developments in nanoscale engineering and molecular design are advancing the field of biomaterials toward bioactive, multifunctional, and targeting materials for applications in tissue Engineering, drug delivery, and imaging [19- 23]. Hydrophobically driven self assembly is a well understood principle that has been shown to facilitate micelle formation. Although quite useful, the library of structures accessible is limited to only a few simplistic geometric configurations that are suboptimal for complex applications. It is believed that other physical phenomena like hydrogen bonding and electrostatic interactions can be exploited to complement hydrophobic interactions allowing for the design of structurally complex, aggregated micelles. Over the past two decades, the fundamental thermodynamic principles that govern micelle formation have been characterized [24-27]. This work has yielded useful tools like the critical packing parameter which can be utilized to predict first order micellar structures making it much easier to create simple geometries such as spheres and cylinders [28]. While useful, simple micelles are quite limited in their adaptability, functionality, and stability which has prompted further research into the development of more architecturally complex micellar structures [29-31]. Recently, twisted and helical micelles have been fabricated demonstrating the feasibility of accessing new structural domains [32-34]. Understanding the structure−function relationships that govern these novel architectures would allow for the rational design of novel peptide amphiphile micelle systems capable of carrying out a variety of complex task besides using hydrophobic driving forces to form self assembled materials. Pharmaceutical micellar and SNEDDS are usually formulated as oil + surfactant and/or co-surfactant/co-solvent mixtures. These systems are diluted with water in vivo or before administration. Micelles and SNEDDS show physical stability in terms of agglomeration or separation of the dispersed phase. These systems also have lower dispersed phase size (≤ 100 nm) giving them transparency. Also, these dosage forms allow the drug to be formulated as both ready-to-use aqueous solutions and as non-aqueous concentrates. The concentrate may be a micellar solution or SNEDDS, which is diluted with water immediately before administration, or administered as it is and gets diluted with gastric fluids in vivo. SNEDDS and micellar systems offer further advantage in that significantly reduced energy requirement for their preparation such that simple mixing is enough for their formation. But, the use of SNEDDS and micellar systems is limited by their drug loading capacity and the usage level of excipients. Surfactants and cosolvents can be toxic at high doses and may be limited in their daily and per-dose uptake levels. In addition, micelles and SNEDDS can be metastable with respect to drug solubility and show drug precipitation upon dilution or crystallization over a period of storage. In vivo drug precipitation upon dilution in stomach can lead to failure in bioavailability enhancement and compromise the competitive advantage of this dosage form. In vitro drug crystallization in a micellar solution or SNEDDS could be very slow and dependent on temperature and handling of the formulation. Improvement in the oral bioavailability of hydrophobic cyclic peptides, like cyclosporine a, using SNEDDS have also shown promise in improving the oral bioavailability of hydrophilic linear peptides and proteins. Improved oral bioavailability from the SNEDDS was also shown for the linear water-soluble nonapeptide leuprolide acetate [35] and dipeptide Nacetylglucosaminyl- N-acetylmuramic acid [36]. Also, intra-gastric administration of SNEDDS of epidermal growth factor is more effective in healing acute gastric ulcers in rats as compared to both intra-peritoneal and intragastric aqueous solution administration [37-38]. Of more direct clinical relevance, stress ulcers are associated with a change in the lipid profile of the gastric mucosa, [39] while each of the barrier breakers displays some affinity for surface active phospholipid (SAPL). Bile salts which can form micelles form a chemical complex with SAPL and ethanol is a solvent for SAPL, while non-steroidal anti-inflammatory drugs (NSAIDs) inhibit the production of prostaglandins controlling SAPL synthesis [40]. Many animal studies have now been reported in which Mucosal protection has been derived from exogenous surfactant administered in various forms, including a commercial grade of lecithin [41] and others [42-43] using a variety of challenges. One of the more interesting avenues is dietary SAPL because this could be useful clinically for preventing ulcers, especially in NSAID patients. Dietary SAPL is available to some degree in milk, which has a modest sprotection rate [44]. Thus the concept of a gastric mucosal barrier of tightly packed hydrocarbon chains provided by surfactant molecules bound to the surface by their polar ends offers a simple physical model for an 'inside lining' based on principles well accepted in the physical sciences, [45-47] together with an explanation for the hydrophobic nature of the normal gastric mucosa and many aspects of mucosal protection. Gastric ulcer results when some aggressive factors are not balanced by the defensive action of some endogenous factors [48-50]. Hence, there is a great need for safe, economic, and efficient antiulcer agents. Natural products emerged as an interesting source of compounds with potential antiulcer activities. The anti-inflammatory activity of naringin has been attributed to the suppression of tumor necrosis factor-alpha, interleukin-6, caspase-3, and nuclear factor kappa-light-chain-enhancer of activated B cells in macrophages [51,52]. Polymeric micelles have been emerged as a successful approach for the site-specific delivery of various drugs. Upon dilution, polymeric micelles are highly more stable than surfactant micelles mainly due to a relatively low critical micelle concentration (CMC) of the former [53]. Therefore, they have been widely employed to deliver chemotherapeutic agents, such as docetaxel alone and in combination with other drugs [54-56]. As well, antitumor activity of doxorubicin has been considerably enhanced through different micellar nanoparticles [57- 59]. Pluronics represent a class of block copolymers formed of hydrophilic blocks of poly(ethylene oxide) (PEO) and hydrophobic poly(propylene oxide) (PPO) in tri-block arrangement of PEO–PPO– PEO (Figure 1) [60]. Pluronics form micelles with a hydrophobic PPO core within a hydrophilic PEO shell in aqueous solutions above CMC. Pluronic nanomicelles showed a noticeable improvement in stability, solubility, biodistribution, and pharmacokinetics of encapsulated drugs. It has been reported that nanoscopic particles showed an increased disposition in inflamed tissue as ulcerative colon of rats being 5- to 6.5-fold higher than in the healthy control [61]. The author suggests that the selective accumulation in the ulcerative areas and the surrounding tissue may be due to either the increased sticky mucus secretion or the particles uptake into the macrophages highly present in the inflamed tissue. The increased residence time at the inflammation sites would permit higher therapeutic effectiveness as well as subsequent dose reduction and cost-effectiveness particularly on large scale. Naringin–PF68 micelles are dispersed spherical particles with nanoscopic diameter 100 nm and narrow size distribution suggesting prolonged circulation times and facilitated access to cells and tissues. The micelles provide extended release up to 10 h for free naringin in different pH release media. These nanomicelles potentiate naringin cytoprotection against ethanol-induced ulcer in rats with dose reduction as reflected by minimized mucosal damage and oxidative stress. Polymeric micelles [62] might be represented as a promising nano-carrier of the phytopharmaceutical naringin with prolonged release as well as enhanced antiulcer encouraging their clinical investigation as alternative of the currently available treatment regimens of ulcer that exhibits some side effects. Peptide Amphiphile Micelles (PAMS) are a class of peptide-based biomaterials consisting of bioactive peptide head groups conjugated to hydrophobic alkyl tails which self-assemble in aqueous solution into micellar structures. They are used as vaccine delivery vehicles that induce a peptide specific antibody response they provide convincing evidence that heterogeneous micelles can enhance lymph node co-delivery of antigens and adjuvants leading to the dramatically improved antibody response observed. Certain physical properties of PAM vaccines including size and charge greatly influence their efficacy. Specifically, spherical and short cylindrical PAMS tens of nanometers in size with near neutral surface charge were found to best enhance antigen immunogenicity. Previous studies have shown that can be utilized to improve subunit vaccine efficacy [64-68]. Peptide amphiphile micelles have been widely utilized as peptide delivery vehicle in a variety of areas such as regenerative medicine, cancer therapy, and vaccination [69]. Both 28 amino acid neuropeptide amphiphiles readily form micelles and show to possess unique anti-inflammatory effects [70]. By combining VIP with the micelle platform, anti-inflammatory biomaterials create considerable potential for limiting transplantation rejection and treating autoimmune disease. Highly positively charged molecules can facilitate greatly enhanced cell uptake because of electrostatic attraction but are often accompanied by deleterious effects such as off target association and significant toxicity [71-74]. On the other hand, highly negatively charged molecules may create electrostatic repulsion with the lipid bilayer, inhibiting their internalization [75]. Therefore, neutral or modest surface charge is preferred in order to facilitate appropriate antigen presenting cell association and internalization [76,77]. Specifically, peptide based nanomaterials have gained considerable interest due to the design flexibility and structural diversity that they provide [78-81]. Surfactants can migrate to surfaces and solvent interfaces, and can also aggregate into micelles, lamellar bilayers, vesicles, hexagonal and cubic mesophases, and other self-assembled supramolecular structures at low concentrations. However, not all amphiphiles display these characteristics. Variables such as the balance of lipophilic and hydrophilic strength of the non-polar and polar moieties, the critical packing parameters, and the composition and geometry of the components, all influence the nature of the selfassembled macrostructure for a given amphiphile [82,83]. Peptides have a range of interesting and useful biological activities but they generally make poor therapeutic agents. The serum half life for intravenous-delivered peptides is often very short. Self-assembled nanoparticles have also been employed to protect bioactive peptides from hydrolytic and proteolytic degradation in-vivo. One opportunity to improve the in vivo half-life of peptides is to have them self-assemble into nanoparticles, which will have different physical properties to the free peptides. In addition to this drug delivery application, selfassembled peptides also have potential use in applications such as platforms or scaffolding for tissue engineering and biological surface engineering [84,85]. One way to control peptide self-assembly is to construct peptide amphiphiles [86]. Surfactant peptides have been constructed using a combination of charged amino acids (e.g. Asp, Glu, Lys, and His) for the polar head-groups, and hydrophobic residues (e.g. Ala, Val, Leu) for the tails. Alternatively, peptides have been conjugated to lipids to form peptidic prodrugs with amphiphilic characteristics such that they self-assemble into a range of interesting nanostructures. Thus, the peptide amphiphilic micelles like SNEDDS are apparently less toxic to the gastric mucosa but stability and scale up issues are of concern [87,88]. The objective of this study was to develop and examine the extent of gastro-intestinal toxicity developed by diclofenac sodium SNEDDS preparation in comparison to diclofenac sodium plain suspension with saline as control administered to rats orally.

Figure 1: Solubility of DFS in individual oils.
Materials
Medium chain Triglycerides (MCT), soyabean oil (SBO), triolein (TRIO), ethyl linoleate (ET), ethyl oleate (EO) isopropyl myristate (IPM), cotton seed oil (CO) was purchased from Sigma, St. Louis, MO, USA; labrasol (Lbsol) was a gift sample from Colorcon laboratories, Goa; Tocopheryl Polyethylene glycol succinate 1000 (TPGS) was gift sample from Eastman chemicals Ltd UK; oleic acid (OA), Polyethylene glycol (PEG) 200, procured from Merck, Mumbai, India; Span 80, lecithin (Lec), was purchased from Sigma, St. Louis, MO, USA; Diclofenac sodium was gift sample from Matrix laboratories, Hyderabad, India; dialysis membrane (10A°) was purchased from Hi Media Laboratories Ltd., Mumbai, India. Centrisart filters (mol wt cut off 20000 Da) were purchased from Sartorius, Goettingen, Germany. All other chemicals used were of analytical grade and solvents were of HPLC grade.
Methods
Solubility study: The oils selected for determining the solubility such as MCT, SBO, OA, TRIO, ET, EO and IPM. To 5 g of oil, DFS was added and the contents were dissolved in methanol/chloroform (1:1) mixture. The solvent mixture was vortexed for 1-3 minutes. The homogeneous solvent mixture was transferred to a 100 ml evaporating flask of rotary evaporator (Buchi, Switzerland). The solvent was vacuum evaporated at 50°C and 57mbar. The oily solution saturated with DFS was allowed to stand at RT(32°C) for 24 h and DFS crystals formed in the oil were separated by ultracentrifugation of oil at 100,000 rpm (micro centrifuge Sartorius, USA). The oil solubility of DFS was determined by, analyzing the DFS content in oil phase using UV spectroscopic method.
Preparation of SNEDDS: Oils, surfactants and the drug were dissolved in 1.5 ml of 1:1 mixture of methanol and chloroform in clean and dry 15 ml culture tube. The remaining ingredients i.e.; hydrophilic surfactants and co solvents were dissolved in another 1.5 ml of 1:1 mixture of methanol and chloroform. Both the solutions were transferred to 100 ml round bottom flask. The oil solution was flash evaporated at 50°C at 110rpm for 20 minutes. The oil solution was finally transferred into a screw capped bottle.
Characterization: The SNEDDS dispersions of diclofenac sodium were characterized for various parameters as described below:
Dispersibility test: The SNEDDS was dispersed into 100 ml of biorelevant media at 37˚C ± 0.5 to assess the self emulsification capacity. It was considered excellent when a visually uniform dispersion was obtained within 1 min and transparent. It was considered good when the time taken for dispersion was 2 min and that the dispersion was milky white. It was considered medium when the dispersion was obtained above 2 min and that it was dull milky white in color with globule size in the micrometer range. Finally, it was considered poor when it took more than 5 min for dispersion and that the resultant system had more dull milky appearance with globule size in micrometer range and that the distribution was non uniform. The SNEDDS which on reconstitution yielded nano-dispersions were further categorized into few classes. The one which spontaneously dispersed yielding a bluish transparent was graded as the “best” whereas the other when spontaneously dispersed but yielded hazy colloidal preparation without any bluish tint was graded as the “good”.
Microscopy: The preparations were observed at a magnification of 450X using epifluorescent microscope (Eclipse E 600, Nikon, Japan) to confirm the presence of oil globules, shape and type of oil droplets. This was done to observe the presence of any oil globules in micron range.
Size and Zeta potential: Size and size distribution was determined by photon correlation spectroscopy (PCS) using Zetasizer 3000HSA/ Zetasizer ZS-90 (Malvern Instruments, Malvern, and Worcs, UK). Each sample was diluted to a suitable concentration with corresponding bio relevant media. Analysis was performed at 25°C with an angle of detection at 90°. The mean size and zeta potential with standard deviation (±SD) was directly obtained from the instrument.
Percent entrapment: The percent entrapment of diclofenac sodium was determined by measuring the concentration of free drug in the dispersion medium. Centrifugal ultra filtration was carried out using Centrisart I (Sartorius AG, Gottingen, Germany) which consists of filter membrane (molecular weight cut-off, 20000 Da) at the base of the sample recovery chamber. The unentrapped drug in the aqueous phase was estimated. Total drug was analyzed after digesting the system with methanol. The percent encapsulation was calculated as follows.
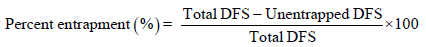
Release studies of DFS SNEDDS were run in triplicate at 37 ± 0.5°C using the USP II dissolution apparatus (Electro lab, Mumbai) at 100rpm. The composition and method of preparation of all the biorelevant media were followed accordingly from USP/NF 2002. At the beginning of each experiment, size 0 hard gelatin capsules were filled with SNEDDS. The capsule was then placed in 500 ml of respective biorelevant media (0.1N Hcl, phosphate buffer, SGF, SIF, and FESIF). Capsules were held at the bottom of the vessel using stainless-steel sinkers. Initially, the release medium was scanned spectrophotometrically from 900 to 200 nm at a gradient rate of 1.5-5 scans/min (Thomson UV spectrophotometer, UK). At the end of each experiment, the release medium was visually examined for signs of turbidity or sedimentation and was judged as transparent, translucent, turbid, or milky. In the fasted state (FASGF, FASIF) resting volumes are quite low, and have been estimated to be about 25 ml [89-91]. However, when a dosage form is administered, some fluid is usually co-administered. In pharmacokinetic studies, this volume is often in the 200-250 ml range. Assuming secretions at a rate of just under 1 ml/min, about 50 ml secretions are expected within 1 h, the longest period during which a fast disintegrating immediate release dosage form is expected to be totally emptied from the fasted stomach [92]. Thus, a realistic volume to simulate the total fluid available in stomach to dissolve simple dosage forms during gastric residence that empty with the fluid after administration in the fasted state would fall in the range of 250-300 ml. It is of note that in the USP dissolution apparatus II, often used for immediate release dosage forms, the minimum volume that can be used is slightly more than 300 ml. Otherwise, the paddle is not completely immersed in the dissolution medium. Based on the above theory, the volume of FASGF and FASIF dissolution mediums in the current studies were set at 500 ml. Samples of 5 ml were withdrawn at preset time intervals. Immediately after the sample removal, the contents were replenished with 5 ml of fresh medium. DFS content in such samples was analysed UV spectrophotometrically at λmax 276 nm.
Gastric Ulcer Studies
Study protocol
The aim of the experimental design was to minimize experimental variables and to avoid bias. All experiments were performed using protocols approved by the institutional animal ethical Committee of UCPSC, Kakatiya University, Warangal.
Animals
Wister female rats were obtained and housed in a temperature and humidity controlled, certified Animal Care Facility. Animals were maintained on wire floor cages over absorbent paper and acclimated to a 12 h day–night cycle for at least one week before experiments. Rat chow (Harlan Teklad, Indianapolis, Indiana) and water were provided ad libitum. At the time of treatment, the rats were 4 months (young) of age.
Study design
In vivo studies were performed to evaluate the ability to prevent DFS related gastric toxicity. A total of eighteen female Wister rats were selected for the gastrointestinal toxicity studies. The selected rats were maintained on a uniform diet and it was ensured that none of them had given any drug at least one week prior to the study. They were divided into two sets. The first set in acute toxicity study comprised of three groups each comprising three rats. The first group was given saline as control, the second group plain diclofenac sodium suspension (Diclofenac sodium powder was dispersed in 1% w/v sodium carboxy methyl cellulose in normal saline at a dose of 25 mg/kg) and the third group was administered DFS SNEDDS preparation (25 mg/kg). A single rat receiving water was kept as normal control. After oral administration, the animals were sacrificed after 4 hrs. Similarly, the second set in chronic toxicity study comprised of three groups. The first group was given saline as control, the second group diclofenac sodium suspension (25 mg/ kg) and the third group DFS SNEDDS preparation (25 mg/kg). On similar lines, after oral administration the rats were sacrificed after one week. All formulations were given orally and studies were conducted in conformity with the animal ethics guidelines for care and use of laboratory animals and approved by ethics committee. For evaluation of the type of lesions in the esophagus and gastric mucosa, a criterion was established. Each tissue sample was scored based on found lesions, according to an arbitrary grading system. The ulcer index (U.I.) for each stomach and esophagus was the sum of scores of all lesions and reported as median (minimum, maximum). The significance of differences between groups was assessed and p<0.05 versus control was taken as significant.
Evaluation of gastric ulcers
When the duration of the study was completed, the animals in acute study were sacrificed after 4 hrs and those in chronic study were sacrificed after one week. Animals were terminally anesthetized with chloroform and the stomach and small intestine were immediately excised and used for analysis.
Quantification of gastric hemorrhagic ulcer area and histological study: The stomachs and intestines of rats after acute and chronic studies were fixed by adding 10% formalin solution and digital images were taken by photo scanning. The ratio of gastric hemorrhagic ulcer area and total gastric area was calculated by the Image- Pro plus (IPP) 6.0 software from Media Cybernetics (Bethesda, CA, United States). The specimens were embedded in paraffin, sectioned and stained with hematoxylin and eosin. Pathological changes were identified under a Nikon Eclipse SOi light microscope (Nikon Co., Japan). To assess gastric mucosal damage score, stomachs were removed, opened along the greater curvature and examined for lesions in the glandular part under a dissecting microscope. The mucosal damage was assessed in a blinded manner by calculation of ulcerogenic index. Essentially, the severity factor was defined according to the length of the lesions, 0: absence of lesions; lesions - 1 mm; 2: lesions 2–3 mm; 3: lesions >3 mm. Stomachs were inspected for the presence of lesions prior to fixation, since gastric ulcers are easier to see in unfixed tissue. The number of ulcers was counted without knowledge of animal treatment.
Statistical analysis: All values were expressed as the mean ± SD. Statistical differences were evaluated by Fisher’s Least Significant Difference (LSD) test followed by a Dennett’s T3 multiple comparison tests. A P value less than 0.05 was considered statistically significant.
DFS solubility studies
The solubility of DFS in individual oils and their combination were determined by UV spectroscopy. The solubility of DFS was high in oleic acid followed by MCT (Figure 1). Since oleic acid had highest solubility of DFS, MCT was combined with oleic acid to find out whether any synergistic effect is achieved with such combination. Interestingly a synergism in DFS solubility was observed. To find out the influence of stoichiometry of these ratios on synergistic solubility of effect of DFS, the ratios of OA:MCT were varied from 1:9, 2:8, 3:7, 4:6, 1:1, 6:4, 7:3, 8:2 and 9:1 respectively. Stoichiometry of the ratios showed synergism and 1:9 & 1:1 ratios exhibited the highest improvement in the synergistic solubility of DFS. These oils were selected to develop oral SNEDDS formulation.
Size and Zeta potential
The size of the nanodispersion of DFS in various biorelevant media ranged from 101-152 nm (Figure 2), zeta potential from 8.4-18.5mv (Figure 3) and percent entrapment from 60.1-90.6 with not much significant variation. The maximum entrapment was observed in FESIF. In 0.1N Hcl low entrapment was observed (Figure 4).

Figure 2: Globule Size of nanodispersions.

Figure 3: Zeta Potential of nanodispersions.

Figure 4: Entrapment Efficiency in Different biorelevant release media.
Release Studies
The initial drug release of the drug was rapid in FASIF followed by phosphate buffer and the total drug release was high i.e.; 74.18% in FASIF followed by phosphate buffer i.e.; 70.14%. In all the media the drug release was seen till 30 hrs and the other important point to be noted is that the entire drug release could not be seen in all the biorelevant media. The drug released for 3-4 hrs approximately in all the media for 50% of release to occur except in SGF and FASGF in which the total percent release itself was 50% till 30 h (Figure 5).

Figure 5: Release profiles of diclofenac sodium from SNEDDS in biorelevant release media.
As shown in Figures 6 in acute toxicity studies, the rats were sacrificed after 4 hrs and then the stomach was isolated and examined visually and microscopically. No lesions or erosions could be seen in the control group I which was administered with saline clearly indicating that the animals were free from ulcers (Figure 6A). In group II (Figure 6B) the animals administered with plain diclofenac sodium suspension were found to have ulcers with erosions in the antrum of the stomach. All the three rats had lesions in combination with erosions in all parts of the stomach but more prominently in the lower portion of the stomach.

Figure 6: A. Rat stomach is an acute study treated with saline (lesions: 0 mm); B.Rat stomach in acute study treated with plains DFS suspension (lesions: >3 mm); C. Rat stomach in acute study treated with DFS SNEDDS (lesions: 0 mm).
Furthermore, different kinds of lesions had developed concurrently. However, in group III the animals which were administered with DFS SNEDDS, ulcers or lesions could not be seen or might be insignificant and thus are protected from ulcer development to a major extent (Figure 6C). As shown in Figure 7, in chronic toxicity studies, the rats were sacrificed after one week and then the stomach was isolated and examined visually and microscopically. No lesions were seen in the stomach of group I (control) revealing the absence of ulcers (Figure 7A). In group II (Figure 7B) severe burns with lesions (ulcers with erosions in the antrum of the stomach) were developed in all the three rats. The location, number, size, and histological features of the lesions were more severe and different to those in group II of acute study. In group III (DFS SNEDDS) in one animal the gastric lesion was a single ulcer with histological features less to those of chronic ulcers (Figure 7C). The findings showed that in both acute and chronic studies with DFS suspension, the gastric mucosa of the animals was not protected from the ulcer causing effects of diclofenac sodium. Among the animals administered with DFS SNEDDS, there was significant protection of the gastric mucosa. To further examine the morphological lesions, we performed the conventional staining. Histological examination showed that the epithelial lining was intact, and the glandular cavity was clear and without any inflammatory cell infiltration in the gastric mucosa in rats treated with DFS SNEDDS (Figure 8A-D). With DFS suspension treatment, marked histological changes were observed, including infiltration of neutrophils, lymphocytes and many erythrocytes into the mucosa. Some secretory glands were broken and many deciduous cells were observed in the glandular cavities (Figure 9A-D). Even mucosal desquamation was found in some gastric tissues. Among the rats with saline treatment, the histological lesions were almost intact (Figure 10A-D). All the results were consistent with the macroscopically changes described in the previous research studies. For rats receiving DFS SNEDDS, the macroscopic morphology was maintained to some extent. However, for rats receiving free DFS, the structural morphology was completely altered (complete loss of gastric mucosal surface), with evidence of bleeding hemorrhagic focus, as well as ulcers. In some cases, all mucosal surfaces showed to be edematous with inflammatory infiltrates. Histological analysis of stomach corroborates the decrease of DFS related toxicity in the stomach mucosa when DFS was administered in SNEDDS dosage form. High amounts of infiltrated red cells were observed for free DFS, while with DFS SNEDDS, these infiltrations were very low. Moreover, the latter showed no evident hemorrhage and the maintenance of gastric pits was observed, which is a sign of substantially reduced DFS toxicity. The same pattern was observed for the esophagus, although to a lesser extent. Complete loss of the epithelium was observed for all rats receiving DFS, while in rats receiving DFS SNEDDS, this effect was slightly decreased. Results provide evidence of cause effect relationship between DFS uptake and gastric injuries, observed by the absence of damage in the control group, and that SNEDDS could offer an advantage of protection against NSAIDs related gastric toxicity. However, it should be noted that the DFS SNEDDS group presented animals without any lesion both in the stomach and the esophagus, contrarily to the DFS group, which is indicative of the gastroprotective potential of this formulation. Prostaglandins (PGs) are important proinflammatory factors with complicated functions in the body. However, some PGs, such as PGE2 and PGI2, are critical in the mucosal defense of the stomach [93] and play a key role in protecting gastric mucosa from injury in the development of Gus [94,95]. NSAIDs cause gastric mucosal lesions, which may be explained by inhibiting the activity of cyclooxygenases (COXs) and thus reducing the production of PGs [96]. The results of the present study showed that DFS SNEDDS resulted in more protection and less injury to the stomach than DFS suspension. This was indicated by the significant decrease in TNFα, iNOS and MDA levels and a significant increase in PGE-2 and GSH levels and GR, GPx, catalase and SOD activities than DFS suspension. So, with regard to the histopathological examination of the stomach and duodenum, DFS SNEDDS treated groups showed less tissue injury (Figure 5). The obtained results of the current study indicated that DFS SNEDDS is more protective of the stomach and duodenum than plain DFS suspension.

Figure 7: A. Rat stomach in chronic study treated with saline (lesions: 0 mm); B. Rat stomach in chronic study treated with plain DFS suspension (lesions: >3 mm & Burns); C. Rat stomach in chronic study treated with DFS SNEDDS (lesions: 0 mm).

Figure 8: Histological assessment of acute and chronic gastric & duodenal mucosal injuries in rats challenged with DFS SNEDDS A. Acute Duodenum tissue section B. Acute Stomach tissue section C. Chronic Stomach tissue section D. Chronic Duodenum tissue section.

Figure 9: Histological assessment of acute and chronic gastric & duodenal mucosal injuries in rats challenged with DFS Suspension A. Acute Duodenum tissue section B. Acute Stomach tissue section C. Chronic Stomach tissue section D. Chronic Duodenum tissue section.

Figure 10: Histological assessment of acute and chronic gastric & duodenal mucosal injuries in rats challenged with saline A. Acute Duodenum tissue section B. Acute Stomach tissue section C. Chronic Stomach tissue section D. Chronic Duodenum tissue section.
Our reports coincide with a study that states that safety concerns associated with non steroidal anti-inflammatory drugs (NSAIDs) report the development of new formulations that minimize adverse events. Nano-formulated diclofenac demonstrate good overall efficacy, prompt pain relief and is well tolerated [97]. Similarly diclofenac submicron particle capsules have been developed using Solu Matrix technology to provide analgesia at lower doses than available solid oral dosing forms. Better pain control was noted across all active treatment groups at 5 hours and pain relief was sustained throughout the treatment period [98]. On similar lines, indomethacin submicron capsules are well tolerated by patients and are a potentially promising treatment option for patients with acute pain [99]. Safety concerns associated with non steroidal anti-inflammatory drugs (NSAIDs) have prompted the development of new formulations that minimize adverse events (AEs) and maintain efficacy [100] like SNEDDS. Indomethacin submicron particle capsules are a potentially promising option for treatment of acute pain [101]. Since in a study Tmax for nanoformulated indomethacin 1.11 ± 0.55 h compared with indomethacin 1.97 ± 0.81 h under fasting conditions, demonstrate faster absorption for the nano-formulated indomethacin and C (max) for nanoformulated indomethacin was higher compared with indomethacin in fasted subjects (3115 ± 900 ng/mL vs. 2759 ± 936 ng/mL) respectively [102] it can be correlated for reduced gastric ulcers of DFS SNEDDS. Since as per the recent reports the Tmax for nano–formulated diclofenac 0.62 ± 0.35 h demonstrate faster absorption than diclofenac 0.80 ± 0.50 h and the Cmax for nano–formulated diclofenac and diclofenac was comparable in fasted subjects (1347 ± 764 ng/mL vs. 1316 ± 577 ng/ mL, respectively) [103] it is obvious that our novel nano–formulated, lower–dose diclofenac sodium SNEDDS demonstrate lower systemic exposure and faster absorption compared with plain diclofenac sodium. Thus, a significant reduction in toxicity of DFS was observed for SNEDDS in gastric mucosa when compared to free DFS with a 10 fold decrease in ulcer index of DFS in acute study and a 6.5 fold decrease in ulcer index of DFS in chronic study (Table 1).
| SNO | Group | Product | Ulcerogenic INDEX | |||
|---|---|---|---|---|---|---|
| Acute Study | Chronic Study | |||||
| Stomach | Oseophagus | Stomach | Oseophagus | |||
| 1 | Test | DFS SNEDDS | 0.3 | 0.1 | 0.5 | 0.3 |
| 2 | Pure drug | DFS IN PB 7.4 | 3.1 | 1.9 | 3.6 | 2.5 |
| 3 | Control | PB 7.4 | 0 | 0 | 0 | 0 |
| 4 | Normal | Water | 0 | 0 | 0 | 0 |
Table 1: Ulcerogenic index in the stomach and esophagus in different groups.
From the solubility study, it was found that the maximum solubility was seen in oleic acid followed by in medium chain triglycerides. A synergistic effect was observed when oleic acid was combined with MCT in various proportions. This synergistic effect could be possibly due to inverse micelle formation in such system and subsequently solubility of DFS into them. The increase in solubility in these systems is directly proportional to the lecithin concentration. The concentration of lecithin in the emulsion is the main factor determining solubility of drugs moderately lipophilic (logP < 2.5), while for more lipophilic compounds the presence of oil is a determinant and for such drugs solubility in submicron emulsion is better than in aqueous lecithin dispersions [104]. It is also shown that the solubilisation capacity strongly depends on the concentration of endogenously secreted materials such as bile salts and phospholipids. However addition of surfactants demonstrated the suitability of SNEDDS to control the precipitation of the drug [105]. The drug:lipid ratio, HLB of the resultant surfactant mixture and production parameters are the factors governing drug release from SNEDDS [106]. Based on this concept, we have optimized systematically the ratios of lipid to surfactants. The importance of biorelevant media in a recent study found that, the dissolution profile of diclofenac sodium from self emulsifying tablets when determined in simulated gastric fluid (SGF) without pepsin was found to be good but not stable. So to get a better in vitro – in vivo correlation, biorelevant media were used. During the process of nanoemulsion formation, a part of the drug may partition into bulk aqueous medium for rapid release, a part remains in the vicinity of oillecithin interface and a part is retained inside the oil. So, our hypothesis of correlating the drug release with its solubility in the oil phase can be explained with a research study which states that the in vitro release of physostigmine is attributed to the retention capacity of the dispersed oil droplets. Increase of the oily phase volume ratio from 20 to 50% did not substantially decrease the rate of release, and decrease of the mean oil droplet size did not affect the release profile [107]. The gastrointestinal toxicity of diclofenac sodium in experimental animals and in humans is well established [108] and is likely to be a limiting factor in the clinical usefulness. The use of SNEDDS for DFS includes reduction of gastric toxicity with minimum exposure in the upper GI tract [109]. It has been confirmed in a study that the increased intestinal permeability, inflammation, bleeding, ulceration are the serious side effects often observed with long term NSAID therapy. So, new formulations of DFS are definitely essential. Diclofenac sodium may appear to exert its GI toxicity through direct contact, distribution to the mucosa upon systemic availability and biliary excretion. In our animal experimental studies, DFS suspension induced toxicity in both the acute and chronic toxicity studies. The observed ulcer effect of the pure drug is likely due to local exposure in the GI tract for a long time. As one of the advantages of SNEDDS is that they do not damage healthy human or animal cells, SNEDDS are suitable for human and veterinary therapeutic purposes [110]. In addition, fine oil droplets empty rapidly from the stomach and promote a wide distribution of the drug throughout the intestinal tract, thereby minimizing irritation. The SNEDDS positively influences drug transport and delivery, along with targeting to specific sites. Interestingly, SNEDDS increases the drug-retention time in the target region, and thus it causes less side effects or toxicities because it does not act on unwanted areas of the body, increases bioavailability and retention time, and decreases drug loss [111]. This may explain the protective effect of DFS SNEDDS on the stomach and duodenum compared to plain DFS suspension. The gastrointestinal adhesion behavior of nanodroplets was examined after oral administration to rats suffering from an experimental gastric ulcer model in order to examine the influence of size on the deposition in inflamed tissue [112]. Highest relative adhesion was found for nanodroplets showing to the relatively small droplets enabling a higher attachment to mucous layers. In gastric ulcer and duodenal ulcers, nanodroplets selectively adhered to inflamed tissue. Inflammation leads to an enhanced mucous production in the affected tissue but the mucous layer in the stomach is thicker than in other regions. Therefore differences from ulcerated tissues in the healthy group became less visible than in colitis where alterations in mucous amount and turnover by the inflammation state have a greater impact on particle adherence. Therefore, the sizedependent deposition of nanodroplets is an offshoot in the development of a new selective drug delivery strategy [113]. An increased adhesion of nanodroplets was observed at thicker mucous layers of inflamed tissue while in ulcerated regions a size dependency was shown. This hypothesis can be related to a research finding in which the usual formulation of DFS may result in retention of drug in the stomach for hours or even days which may cause retarded absorption, gastrointestinal toxicity and delayed plasma peak concentrations. Moreover, diclofenac sodium has been shown to undergo considerable first pass metabolism, limiting its oral bioavailability. The assumption of local exposure of the drug can be supported in a study which assumes that the change of a conventional formulation of DFS is needed to reduce the GI damage. It reveals that the percent incidence of GI toxicity attributed to sustained and enteric coated diclofenac sodium preparations (52.1%) is signicantly greater than that attributed to immediate release tablet preparations (37.5%) because of more residence time and high local exposure of the drug to the GI membrane. In another study, it explains that the mechanisms involved in upper GI damages of some of the NSAIDs, e.g., aspirin, may be due mainly to their direct local effects. So, assessment of diclofenac sodium toxicity on the entire GI tract revealed that there was a particular advantage for using SNEDDS for DFS. With SNEDDS, the local exposure of the drug is minimized, since the drug is either absorbed rapidly due to enhancement in absorption or globule absorption may take place. Our hypothesis could be related to research finding which states that, Gentamicin, an amino glycoside is poorly absorbed from the gastrointestinal (GI) tract. When administered in labrasol micro emulsions to rat small intestine and colon, it facilitates the transmucosal delivery by enhancing paracellular absorption and inhibition of efflux mechanism in the enterocytes [114]. In another report, labrasol with the concentration of 0.1 and 1% is shown to increase the permeability of mannitol by 4.6-fold and 33.8-fold, respectively. The mechanism of opening of tight junctions is found to involve F-actin-related changes and redistribution of ZO-1 [115]. Our findings are further strengthened by a study that vancomycin hydrochloride when formulated with Labrasol and TPGS, the oral absorption is enhanced by 2.2 and 2.4 times. This is a measure for the decreased retention time in the GIT. It is also found that, labrasol increases intestinal absorption and bioavailability of P-gp substrate rhodamine123 by inhibiting the function of P-gp. We hypothesize that SNEDDS allowed the reduction of gastric toxicity by avoiding the direct contact between gastric mucosa and the drug by retardation of its release. Since DFS belongs to the class II drugs of the BCS, this means that the limiting step for its uptake will be the release from the polymeric matrix. The reduced uptake of DFS in the stomach by SNEDDS could, therefore, have a major impact in patients taking chronic therapeutics. The fact that SNEDDS products are biocompatible [116] represents an extra advantage, which was confirmed by the fact that no toxicity non-related to DFS was found. Other polymers have been reported to encapsulate NSAIDs to alter their delivery. As an example, IBU-loaded PLGA nanoparticles [117] and Eudragit L100 nanoparticles [118] allowed the controlled release of IBU. Others reported diethylaminoethyl-dextran IBU nanoparticles allowed a pH sensitive, burst release. Furthermore, nanocapsules with indomethacin effectively prevented intestinal lesions [119]. As intended from the established goals, this study is in fact a proof of concept for a formulation that will allow DFS administration with reduced gastric toxicity. This would ultimately increase DFS efficacy and safety, offering a major advantage over conventional formulations in improving clinical outcome. Furthermore, these results offer the possibility of using DFS SNEDDS for an extended number of drugs of the same group. These studies significantly prove that labrasol enhances absorption transcellularly and paracellularly thereby decreasing the local exposure of DFS in the GIT. Thus, our results clearly indicate that rats administered with DFS SNEDDS in acute and chronic toxicity studies exhibit significantly less macroscopic damage to the gastric mucosa than with plain suspension. This seems reasonable to propose that at least part of the reduced gastric/intestinal toxicity associated with DFS SNEDDS usage reflects the reduced gastrointestinal toxicity which suggests that, this may prove of interest for further evaluation in the clinic.
In summary, our findings revealed that DFS SNEDDS could be prepared by emulsion diffusion evaporation technique. DFS formulated SNEDDS showed the least size of 101 ± 3.5 nm, zeta potential of -18.5 ± 1.6 mv and entrapment efficiency of 90.6 ± 2.1 in FESIF respectively. The drug released for 3-4 h approximately in all the media for 50% of release to occur except in SGF and FASGF in which the total percent release itself was 50% till 30 hrs. A significant reduction in toxicity of DFS was observed for SNEDDS in gastric mucosa when compared to free DFS with a 10 fold decrease in ulcer index of DFS in acute study and a 6.5 fold decrease in ulcer index of DFS in chronic study. Histopathological examination of the stomach confirmed the biochemical and molecular findings of less tissue injury. Further investigations should be undertaken to study accurately the mechanisms of DFS SNEDDS on the stomach wall. It can be concluded that DFS SNEDDS results in more protection and less injury to the stomach and duodenum.
The authors are grateful to University College of Pharmaceutical Sciences, Kakatiya University. This work was supported by Natco Research Center (NRC), sanathnagar, Hyderabad and is sincerely acknowledged. We are grateful to the entire NDDS unit team of NRC for their assistance in the analytical measurements and their efforts are greatly appreciated. We are deeply indebted to Dr. B. Madhava Reddy, Professor & Principal, G. Pulla Reddy College of Pharmacy, Hyderabad for permitting to conduct animal studies. The authors also would like to thank to Dr. V. Birabhadhar Rao, Pathologist, V.B.R Diagnostics, Warangal for his kind technological support in electron microscopy experiments.