Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Review Article - (2013) Volume 2, Issue 1
In eukaryotic cells, DNA damage repair occurs on a template DNA that is organized with histones to form nucleosomes and chromatin structures. As such, chromatin plays an important role in DNA damage repair. In this review, we will use “chromatin damage repair” as a framework and highlight recent progress in understanding the role of chromatin, chromatin modifiers, chromatin binding effectors (e.g., the PWWP domain proteins), and the p53 tumor suppressor. We view chromatin as an active participant during DNA damage repair.
<ARR: Access-Repair-Restore; ATM: Ataxia Telangiectasia Mutated; DDR: DNA Damage Response; DSB: DNA Double-strand Break; HAT: Histone Acetyltransferase; HR: Homologous Recombination; HRP: Hepatoma-Derived Growth Factor Related Protein; MDC: Mediator of DNA Damage Checkpoint Protein 1; MRN: Mre11-Rad50-Nbs1; NE: Nucleotide Excision Repair; NHEJ: Non-Homologous End Joining; 53BP1: p53 Binding Protein 1; γH2AX: phosphorylation of H2AX Ser139.
Chromatin structure and organization
In eukaryotic cells, the nucleosome is the basic structural unit of chromatin. It is composed of a 147 bp of DNA tightly packed around a core histone octamer, which contains an H3-H4 tetramer and two H2AH2B heterodimers [1,2]. With the help of about 20 to 60 base pairs of linker DNA, nucleosomes are further packed into an approximately 11- nm “beads-on-a-string” fiber, which defines the first level of chromatin structure [2]. At the second structural level, 30-nm chromatin fiber is organized by further packing of nucleosome arrays with linker histone H1 [3]. Furthermore, the chromatin fibers are folded into higher order structures by looping and further folding during interphase. At last, chromatin is further compacted into condensed metaphase chromosome during mitosis.
The interphase chromatin can be divided into two categories due to various degrees of chromatin condensation and composition. While chromatin in a less condensed conformation is termed as euchromatin that is usually related to active transcription, heterochromatin is known as gene-poor region with tightly packed chromatin [4]. Constitutive heterochromatin consists of some structural elements, such as, telomeres, centromeres, non-coding and small repetitive regions. Besides, facultative heterochromatin can be converted from euchromatin due to differentiation, X-chromosome inactivation and so on [5]. Although the mechanism of higher-ordered chromatin structure formation is still not completely clear, it is known that the regulation of chromatin structure dynamics is dependent on many factors, including DNA methylation, histone variants, histone modifications, and binding of non-histone chromatin architectural proteins and protein complexes [6]. From yeast to human, regulation of eukaryotic chromatin organization has vast significance in regulating many DNA-dependent cellular activities, such as transcription, replication and DNA damage repair. In the following parts, chromatin structure modulation during DNA damage repair in the mammalian system will be further discussed.
DNA damage response signaling network
Genome stability is critical for biological functions and cell viability. However, genome is continuously under threats from various exogenous or endogenous DNA damaging stresses. External ionizing radiation, UV irradiation and environmental chemicals can cause DNA damages. Internal metabolic products, such as Reactive Oxygen Species (ROS), and spontaneous errors during DNA replication alter the genetic information stored in the DNA double helix [7]. These threats cause several types of DNA lesions, including base damages and mismatches, bulky adduct intra-and inter-strand crosslinks, as well as single and double strand breaks [8].
To counteract several harmful cellular outcomes of DNA lesions, a defense system called DNA damage response (DDR) follows, which is essential for anti-cancer and anti-aging processes [9]. DDR is an integrated network of highly ordered signaling cascades. When facing DNA insults, some protein complexes called “sensors or mediators”, are recruited to DNA damage sites, which can be observed as nuclear foci under microscope. Next, the sensors activate “transducers”, such as a protein kinase ATM/ATR, to transfer and amplify signaling to downstream “effectors”. Many effectors play a key role in deciding the cell fate. In order to survive, cells with transient cell-cycle arrest may resume cell proliferation after successful DNA repair, whereas others may enter permanent cell-cycle arrest and senescence with unrepaired DNA damages. In the worse scenarios, programmed cell death or apoptosis occurs when DNA damage is too severe. Additionally, some other effectors also establish a feedback loop to control the DDR signaling pathways to maintain the homeostasis of cell survival and death after DNA damage [10,11].
As mentioned above, DNA repair is a crucial mechanism to rescue cells from DNA damage stress. According to the different types of DNA damage produced, there are at least six distinct DNA repair pathways evolved to deal with DNA lesions during DDR: Nucleotide excision repair (NER), base excision repair (BER), mismatch repair, cross-link repair and double-strand break (DSB) repair [12]. DSB repair can be divided into two different pathways: non-homologous end joining (NHEJ) and homologous recombination (HR) [13].
DNA double-strand break (DSB) is the most lethal type of DNA damage due to a complete breakage of DNA backbone. Nonetheless, the molecular mechanisms described below for DSB repair are also found to participate in DDR initiated by other DNA damage. DSB repair in mammalian cells is a coordinated signaling pathway initiated with immediate activation of the MRN (mre11-rad50-nbs1) complex and the kinase ataxia telangiectasia mutated (ATM). DSB stressactivated ATM kinase phosphorylates H2AX on Ser139 (also called γH2AX) at damage sites. Then, γH2AX spreads along the chromatin up to hundreds of kilobases from initial site with help of the ATMMRN- MDC1 (mediator of DNA damage checkpoint protein 1) complex [14,15]. This amplified signal recruits massive number of proteins to contribute to the repair process or regulating the cell-cycle checkpoints.
For actual repair, NHEJ is a predominant repair mechanism that directly rejoins the broken DNA ends. Since it utilizes single stranded overhangs at DSB ends, NHEJ can be done at anytime but favored in G1 phase of the cell cycle. Although it is a fast way to response to DSB, NHEJ has higher chance to induce mutagenesis by generating deletions or chromosome translocations. During NHEJ in mammals, Ku70/80 heterodimer with DNA dependent kinase catalytic subunit (DNAPKcs) bind to DSB site for end tethering,then recruit Artemis and DNA polymerases μ and λ for gap filling. Finally, ligation step is triggered by DNA ligase IV complex and scaffold proteins, such as X-ray repair cross-complementing protein 4 (XRCC4) and XRCC4- like factor (XLF) [16,17].
Compared to error-prone NHEJ, HR can guarantee high fidelity of repair but require more steps and more protein effectors. Specifically, the MRN complex recognizes DSB sites and generates single-strand DNA intermediates by CtBP-interacting protein (CtIP) mediated resection during HR [18]. Replication protein A (RPA), RAD51 and RAD52 bind single-stranded ends, and direct synapsis and strand invasion to the homologous template. The DNA synthesis and ligation are completed finally to restore disrupted genetic information [19]. Because HR requires homology searching of sister chromatids as accurate template in error-free repair, it only occurs in S and G2 phases [20].
Dynamics of chromatin structure regulates DDR
Given that DNA is organized into the chromatin structure, it is not surprising that chromatin structure influences multiple steps during DDR. Usually, less condensed chromatin regions are more sensitive to DNA-damaging agents than those with a higher level of compaction [21,22]. In addition to chromatin’s natural barrier function, the dynamics of chromatin organization affects DDR as well, which was first described in a model termed “access-repair-restore” (ARR) (Figure 1) [23,24]. This model elucidates three main steps after DNA damage: (1) the site of damaged chromatin is detected and become more accessible; (2) reorganization of chromatin structure allows processing of DNA repair factors; (3) changes in chromatin organization is restored after repair has finished [25,26]. Previously, the “access” step was always thought that proteins are mainly removed from the chromatin to increase accessibility. Recently, this is renamed as the “prime” step, which involves both adding-in and taking-out of chromatin factors at this initial stage of DDR [27].
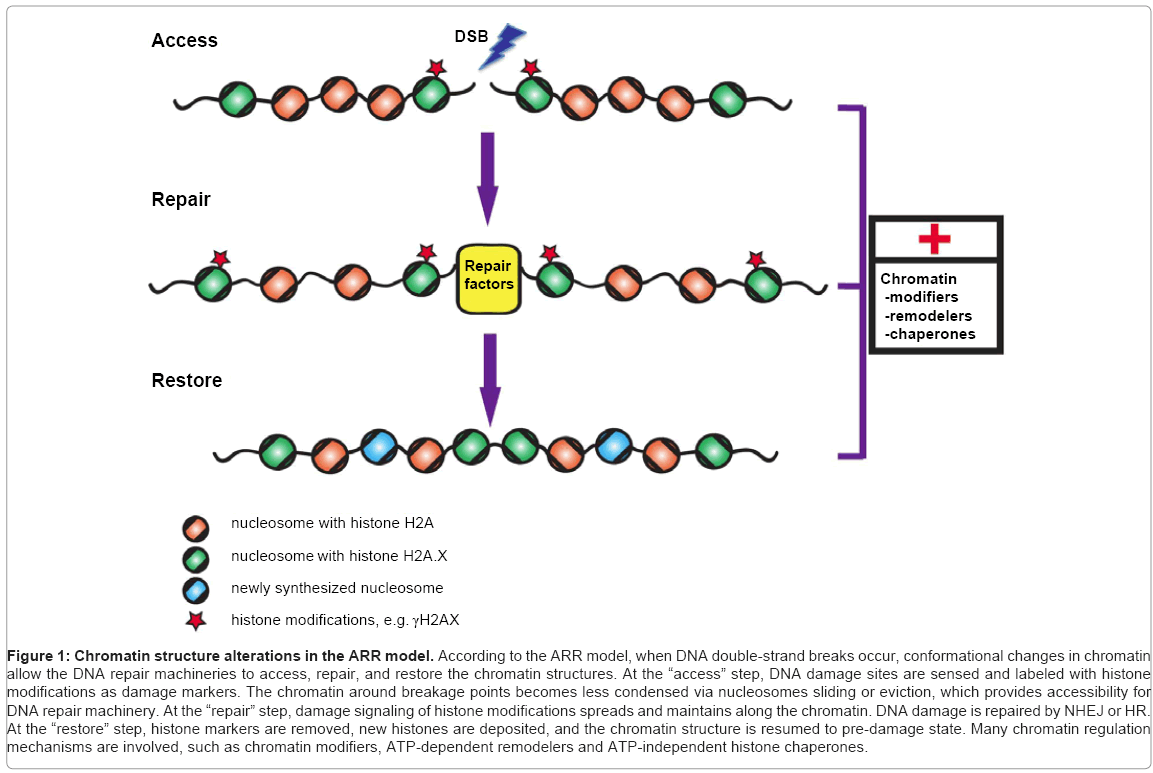
Figure 1: Chromatin structure alterations in the ARR model. According to the ARR model, when DNA double-strand breaks occur, conformational changes in chromatin allow the DNA repair machineries to access, repair, and restore the chromatin structures. At the “access” step, DNA damage sites are sensed and labeled with histone modifications as damage markers. The chromatin around breakage points becomes less condensed via nucleosomes sliding or eviction, which provides accessibility for DNA repair machinery. At the “repair” step, damage signaling of histone modifications spreads and maintains along the chromatin. DNA damage is repaired by NHEJ or HR. At the “restore” step, histone markers are removed, new histones are deposited, and the chromatin structure is resumed to pre-damage state. Many chromatin regulation mechanisms are involved, such as chromatin modifiers, ATP-dependent remodelers and ATP-independent histone chaperones.
In general, there are three major mechanisms involved in the regulation of nucleosome/chromatin organization: post-translational histone modifications, ATP-dependent chromatin remodeling, and ATP-independent chromatin modulation [28]. These three types of machineries function in coordinated manner throughout the ARR process of DNA damage repair.
Diverse post-translational modifications, such as methylation, acetylation, phosphorylation, citrullination, ubiquitylation, sumoylation and poly ADP-ribosylation, preferentially occur at histone tails [29-34]. These histone modifications could alter chromatin structure, by changing histone-DNA and histone-histone contacts or recruiting different chromatin factors [35]. It has been proposed that covalent histone modifications work sequentially or in combination to form a “histone code” and define various downstream biological outcomes like DNA transcription, replication and repair [36]. Recently, more and more published work indicates that DSB repair is a new hot target to decipher stepwise DNA-damage-induced histone code [37].
Poly ADP-ribosylation
As the earliest modification detected at DNA damage sites, poly ADP-ribosylation is produced by poly ADP-ribose polymerases (PARP) and erased by poly ADP-ribose glycohydrolase (PARG) [38,39]. NuRD (nucleosome remodeling and deacetylase) and Polycomb complexes are recruited to poly ADP-ribose (PAR) chains at lysine residues within the N-terminal tail of histones. These chromatin regulators help to temporarily form condensed repressive chromatin, inhibiting any possible DNA breakage from RNA polymerase interruption and further transcription [40]. Intriguingly, chromatin at DSB site also has an increased accessibility by association with ATPase motor containing chromatin remodeler complexes (e.g. ALC1, amplified in liver cancer 1) in PAR-dependent manner [41]. Thus, poly ADP-ribosylation perhaps performs a critical role in immediate damage sensing and pilots downstream DDR events.
Phosphorylation
Phosphorylation of H2AX Ser139 (γH2AX) was first discovered in 1998 and serves as a primary marker of DDR signaling activation [42]. The histone H2AX variant exists in different cell lines with genomic occupancy ranging from 2% to 20% of the total amount of H2A [42]. A ChIP-seq analysis shows that the distribution of H2AX is enriched at euchromatin regions other than heterochromatin [43]. Moreover, H2AX knockout mice are more sensitive to ionizing radiation and show defects in DNA repair and genome integrity maintenance [44].
As mentioned before, the MRN complex binds to DSB sites, recruits and activates ATM. ATM in turn induces γH2AX, providing a binding site for MDC1 [45-47]. The MDC1 protein docks more ATMMRN complexes to promote the propagation of γH2AX markers from the original break points to a region that could be as far as 2 megabases away [14,15,17]. Using 2D and 3D confocal microscopy and ChIP analyses, it is revealed that γH2AX shows high densities near DSB sites and weaker signals at distant sites [48] . Although the mechanism underlying γΗ2AX expansion is not fully understood, it is known that nuclear diffused ATM may trigger long-distance γΗ2AX formation via MDC1-independent manner during V(D)J recombination in pre-B cells [49]. In addition to MDC1, BRIT1/MCPH1 (microcephalin) was identified as a docking protein to recognize γΗ2AX [50]. It can recruit SWI-SNF chromatin remodeling complex to enhance chromatin accessibility [51].
At the final “restore” stage, γΗ2AX must be removed to resume previous local chromatin status. Several protein phosphatases such as PP2A, PP4 and PP6 have been discovered to target γH2AX [52-54]. WIP1 was a recently identified enzyme to dephosphorylate not only γH2AX but also some other ATM/ATR substrates [55]. Since wild-type p53-induced phosphatase 1 (WIP1) is upregulated by p53, there is a p53-mediated feedback loop to restrain DNA damage signal amplification by an increase in γH2AX phosphorylation levels.
In addition to γH2AX, phosphorylation of H2AX Tyr142 serves as an important marker for undamaged status of the whole genome. The phosphorylation of this adjacent site to Ser139 is constitutively maintained by Williams syndrome transcription factor (WSTF) under unstressed conditions, and then is removed by eye absent homologue 1/3 (EYA 1/3), accompanied with γH2AX generation [56,57]. This conserved phosphorylation of H2AX is also important in cell fate decisions in concert with γH2AX.
Acetylation
Histone H4 acetylation is important in forming chromatin structure related to active transcription [58]. Especially, acetylated H4 Lys16 can perturb the ionic interaction between the N-terminal tail of histone H4 and H2A-H2B dimer of an adjacent nucleosome to prohibit further package of the 30 nm chromatin fiber [59,60]. After binding to chromatin via γH2AX-dependent manner, the docking protein MDC1 rapidly recruits a multiple subunit histone acetyltransferase (HAT) complex, NuA4, whose Tip60 subunit acetylates H4K16 [61]. Similar to γH2AX, H4 acetylation also spreads out from DSB sites to a region as far as hundreds of kilobases away. Disruption of Tip60 activity can abolish H4K16 acetylation and promote compaction of chromatin [62]. Conversely, H4K16 acetylation can be removed by histone deacetylases, HDAC1/2, which may prevent Ku complex sliding and dissociation from DNA ends especially in NHEJ pathway [63].
Ubiquitination
Besides the NuA4 complex, MDC1 also serves as a platform for sequential recruitment of ubiquitin ligases RNF8 and RNF168 [64]. On one hand, the RNF8-RNF168 cascade triggers ubiquitination of H2A, H2AX and H2B for chromatin structure relaxation. On the other hand, the ubiquitination is required for binding of other chromatin factors such as breast cancer type 1 susceptibility protein (BRCA1) and p53 binding protein 1 (53BP1) [65,66]. Additionally, H2B monoubiquitination mediated by RNF20-RNF40 heterodimer directly leads to chromatin decompaction at DSB sites [67].
Methylation
Similar to ubiquitination, histone Lys methylation facilitates recruitment of repair factors during DDR. The tudor domain of 53BP1 recognizes H3K79me2, a methylation site that is normally embedded within the body of nucleosomes and becomes exposed upon DNA damage mediated conformational changes [68]. Furthermore, the interaction between 53BP1 and H4K20me2 is enhanced by DSB [69]. Interestingly, to maintain genome integrity, a quite high level of H4K20me2 (>80%) is constitutively present. After DSB, H4K20me2 may be further exposed, or established from unmodified histone H4 with de novo methylation by a methyltransferase MMSET [70,71]. This increasing H4K20me2 level is crucial for 53BP1 recruitment and a cascade of downstream regulatory events.
Besides covalent histone modifications, ATP-dependent chromatin remodeling is another important mechanism for chromatin reorganization. Chromatin remodelers utilize ATP hydrolysis to affect chromatin structure by either replacing histones with corresponding variants, or repositioning (sliding or removal) of nucleosomes [72]. At least three remodeler families have been found to play a role during DSB repair in mammals: Swr1-like, Snf2-like and Rad54-like families [73]. And these chromatin remodelers regulate different stages of DSB repair. For example, ION80 complex, a member of Swr1-like family, is recruited to DSB sites by its ARP8 subunit, and facilitates H2AX phosphorylation expansion by its ARP5 subunit at the “access” step [74,75]. Another example is the human NuA4 complex, which contains Tip60 acetyltransferase and the p400 ATPase motor protein. Once DSB stress occurs, histone H2A is replaced with H2A.Z variant by p400, and H4K16 acetylation is triggered by the TRRAP-Tip60 subunits. Both events result in the formation of relaxed chromatin [76].
Histone chaperones are histone-binding proteins that contribute to both assembly and disassembly of nucleosomes without using the ATP energy. Given their roles in chromatin structure regulation, it is anticipated that histone chaperones are involved in ARR as well. The FACT (facilitating chromatin transcription) complex is a heterodimer of Spt16 and SSRP1 (structure specific recognition protein-1), which is known in the regulation of transcription elongation [77,78]. Its function is inhibited by PARP1 mediated poly ADP-ribosylation during “repair” [79]. At the “restore” step, however, FACT complex replaces γΗ2AX-H2B with unmodified H2AX-H2B or H2A-H2B dimers thereby facilitating the removal of DNA damage markers [80]. In addition, Asf1 (anti-silencing function 1) and CAF-1 (chromatin assembly factor 1) synergize to exchange histones H3/H4 with newly synthesized H3/H4 during the “restore” step. The newly synthesized histone H3 is acetylated at Lys56, a modification important for the nucleosome reassembly process. However, it is rapidly removed by HDACs (hSIRT2/3) to allow chromatin condensation [81,82]. It is still unknown whether histone chaperones capture displaced histones during the initial “access” step [83].
In sum, histone modifications, ATP-dependent and -independent chromatin mechanisms play important roles at all steps of DDR. The crosstalk among these mechanisms is indispensable to trigger spatially and temporally coordinated events for DNA damage repair (Figure 1).
Despite of the primary regulatory machinery of chromatin described above, some chromatin-associated proteins also serve as effectors to modulate DNA damage pathways. The PWWP domain containing proteins were recently found to play important roles in DDR (Table 1). In the following section, we will further discuss the role of this family of proteins serving as chromatin binding factors and DDR effectors.
| Effectors | Homologs | PWWP Domain | Histone methylation recognition |
Histone modifier activity |
DNA damage stimuli |
Role in DNA damage repair |
Role in DDR |
|---|---|---|---|---|---|---|---|
| Pdp1 | Fission Yeast | Residue 52-1124 | H4K20me | Not found | UV, ionizing radiation | Yes | Set9 recruitment (damage signaling transduction) |
| MUM1/ EXPAND1 |
Human, mouse, rat, bovine |
Residue 411-472 | Not found | Not found | Ionizing radiation, neocarzinostatin |
Yes | Recruited by 53BP1; chromatin relaxation (NHEJ repair) |
| MMSET (WHSC1) |
Human, mouse | Residue 222-286; Residue 890-942 |
H4K20me | Methylate H4K20 |
Ionizing radiation | Yes | 53 BP1 recruitment (damage signaling transduction) |
| LEDGF (PSIP1/ p75) |
Human, mouse, rat, bovine |
Residue 1-63 | H3K36me3 (PSIP1/ p52) |
Not found | Ionizing radiation, camptothecin mitomycin C |
Yes | CtiP recruitment (HR Repair) |
Table 1: The role of the PWWP domain proteins in DNA damage reponse.
The PWWP domain in DNA and methyl-histone binding
The PWWP domain is a member of the “Royal family” protein domains, including the PWWP-, Tudor-, chromo- and MBT-domains [84]. The PWWP domain features a conserved Pro-Trp-Trp-Pro core motif. It was firstly identified in the Wolf-Hirschhorn syndrome candidate 1 (WHSC1) protein and was predicted to mediate proteinprotein and protein-DNA interactions [85]. So far, there are more than 60 proteins that have been discovered to have PWWP domain [86]. Since PWWP domain shares significant sequence similarity with Tudor domain and other members, it was postulated to recognize methylated histone residues [84]. The bromodomain and PHD finger-containing protein 1 (BRPF1) binds to a histone H3K36me3 peptide using its PWWP domain [87]. Recently, the Dr. Min’s group systematically studied several representative human PWWP domains in biochemical and crystallization studies (Wu et al., 2011). A typical PWWP domain has three structural motifs: a N-terminal β-barrel, an insertion between the second and the third β-strands, and a C-terminal α-helix bundle [88]. The PWWP domain recognizes histone Lys methylation with an aromatic cage [88]. Meanwhile, the PWWP domain is also known as a non-specific DNA-binding module due to its DNA-interacting surface with basic electrostatic features. It is reported that the PWWP domain of DNA methyltransferase Dnmt3b targets the enzyme to pericentric heterochromatin [89]. The PWWP domain of mutS homolog 6 (MSH6), is another example showing affinity to double stranded DNA during DNA mismatch repair [90].
The HRP subfamily of PWWP-domain containing proteins
The hepatoma-derived growth factor (HDGF) is a nuclear protein, which was initially identified as a protein with mitogenic activity [91,92]. HDGF and other HDGF-related proteins (HRPs), including HDGF-related proteins HRP-1, -2, -3, -4 and lens epithelium-derived growth factor (LEDGF) form a subfamily of proteins that contains the PWWP domain. Members of HRP subfamily share a highly conserved N-terminal region containing the PW(H)WP domain but differ from their diverse C-termini.
As a prototype protein of this family, HDGF is highly expressed in certain tissues during development [93], and the over-expression of HDGF was detected in many human cancers [94]. HDGF protein interacts with C-terminal binding protein (CtBP) via a “PKDLF” motif within the PWWP domain to repress the SET and MYND domain containing 1 (SMYD1) gene expression [95]. Although most PWWP domain-containing proteins bind to DNA via non-specific manner, it is found that HDGF associates with a 37 bp DNA showing certain degree of sequence conservation on the SMYD1 promoter through its PWWP domain [96]. LEDGF is the alternative splicing isoform p75 of PC4 and SF2 interacting protein 1 (Psip1) [97]. It consists of a N-terminal PWWP domain, an AT hook-like motif and a C-terminal HIV integrase binding domain (IBD) [98,99]. LEDGF can bind to HIV- 1 integrase via its IBD. This prevents HIV-1 integrase from degradation by proteasome, and bridges viral genome integration to the host genome [100,101]. LEDGF is also shown to bind to MLL (myeloid/ lymphoid or mixed-lineage leukemia), an H3K4 methyltransferase, via Menin in transformed myeloid progenitors [102]. A short splicing isoform p52 of Psip1 is able to recognize H3K36me3 specifically via its PWWP domain and regulates alternative splicing [103]. Furthermore, HDGF2 (alias of HRP-2), which also contains a C-terminal IBD, plays an accessory role in LEDGF-mediated HIV integration [104]. Although extensive literature documents studied the potential biological function of HDGF and LEDGF, biological functions of HDGF2 and other members of this protein family remain still largely unknown.
PWWP domain-containing proteins in DDR
The PWWP domain-containing proteins function as chromatinassociated factors and contribute to various biological events using DNA as a template, such as transcription and DNA replication. How the PWWP domain-containing proteins are involved in DNA repair process is a field of increasing interest. Four proteins with PWWP domain were shown to regulate DDR at various steps.
The PWWP domain-containing protein (Pdp1) protein in fission yeast is a PWWP protein with no clear homologous protein found in higher eukaryotes [105]. Pdp1 prefers to recognize H4K20me1 via its PWWP domain and facilitates histone methyltransferase Set9 to further produce di- and tri-methylation of H4K20. Pdp1 is also critical for Crb2 (53BP1 homologous in yeast) accumulation and phosphorylation under DNA damage stress [105]. Depletion of Pdp1 disrupts DNA repair pathway and makes yeast more sensitive to DNA damage treatment [105]. Moreover, the Pdp1 PWWP domain was found to bind both H4K20me3 peptides and double-strand DNA fragments simultaneously in biochemical and crystal studies [106].
The human melanoma associated antigen mutated 1 (MUM1/ EXPAND1) shares homology with proteins in mammals (mouse, rat and bovine) [107,108]. The PWWP domain of EXPAND1 locates at its C-terminal region. Although no histone methylation binding affinity has been found yet, EXPAND1 is a chromatin-bound factor recruited to damage sites by interacting with 53BP1 and enhances further chromatin decondensation upon DNA damage stress [107,108].
MMSET (also called WHSC1 or NSD2) containing a SET domain, has been studied to generate several histone methylations (H3K4me3, H3K27me3, H3K36me2, H3K36me3) and regulate transcription [109-113]. However, its function in DNA repair regulation was discovered only recently [70]. MMSET is phosphorylated by ATM and then accumulated at DNA damage sites depending on the H2AX-MDC1 protein complex. As a SET domain-containing histone methyltransferase that harbors a PWWP domain, MMSET induces di and tri-methylation of H4K20 accumulated at DSB sites and facilitates recruitment of 53BP1 [70]. Strikingly, MMSET seems to mediate crosstalk among multiple modifications, including phosphorylation, ubiquitination and methylation, during DDR [70].
Most recently, the Jäättelä group reported that LEDGF (Psip1/ p75) plays an important role in DSB repair in addition to its wellknown functions, such as promoting HIV integration and modulating alternative splicing [114]. LEDGF helps to defend genotoxicity and protect survival of cells under DNA damage by enhancing the recruitment of CtIP at DNA break sites especially [114]. Deficiency in LEDGF results in impaired DNA end resection during HR repair.
To sum up, these PWWP proteins bind to chromatin and maintain genome stability in physiological conditions. Once DNA damage occurs, they react immediately to promote recruitment of DNA repair factors like 53BP1 and CtIP, and regulate chromatin condensation during DDR signaling (Table 1). It is anticipated the underlying correlation between the PWWP domain and DNA repair mechanism will be studied further with more identified PWWP proteins.
Unlike less-characterized PWWP proteins, p53 is much more versatile as a “molecular node” that effects in multiple facets of DDR [115,116]. p53 was first discovered as an oncogene in 1979, but was later recognized as a tumor suppressor protein at the hub of an intriguing signaling network in mammalian cells [117-119]. Structural studies found that p53 contains an N-terminal transactivation and prolinerich domain (residues 1-92), a DNA binding domain (residues 100- 300), a tetramerization domain (residues 307-355), and a C-terminal regulatory domain (residues 356-393) [120]. It is reported that p53 gene is altered in more than 50% of human cancers, and the mutations predominantly occur within its conserved DNA binding domain [121,122]. Since tumor development always leads to accumulation of genetic DNA lesions, p53 plays a very critical role in guarding genome stability in DDR and preventing tumorigenesis [123,124].
Activation of p53 upon DNA damage stress
Under unstressed conditions, murine double minute 2 homolog (MDM2) exerts E3 ubiquitin-protein ligase activity to ubiquitinate p53. p53 upregulates MDM2 transcription to establish a negative feedback loop [125,126]. In addition, MDM4 (also known as MDMX) is identified in a heterodimer with MDM2 to facilitate p53 ubiquitination, and MDM4 alone can repress p53-mediated transcription [127,128]. p53 turnover is also controlled by other proteins such as DAXX (deathdomain associated protein) and HAUSP (herpersvirus-associated ubiquitin-specific protease), which reduce MDM2 auto-ubiquitination and further enhance p53 degradation [129,130]. In this way, p53 is kept at very low levels in a latent form in cells without DNA damage stress [128,131,132].
When DNA damage stimuli are detected, activation of p53 can be achieved by several steps (Figure 2) [120]. First, p53 is stabilized by decreasing its ubiquitination mediated by the MDM2 pathway. The phosphoatidylinositol-3 kinase-like kinase family members, ATM and ATR (ataxia-telangiectasia and Rad3-related) phosphorylate MDM2 and disrupt MDM2-DAXX-HAUSP complex thereby inducing p53 accumulation by preventing its ubiquitination [133].
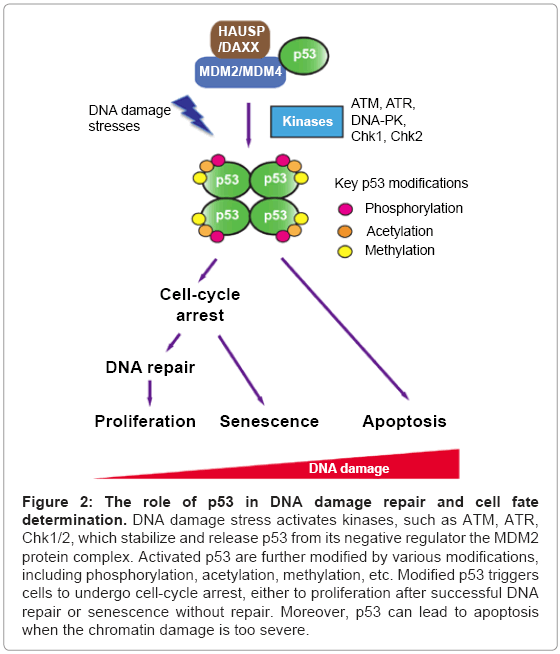
Figure 2: The role of p53 in DNA damage repair and cell fate determination. DNA damage stress activates kinases, such as ATM, ATR, Chk1/2, which stabilize and release p53 from its negative regulator the MDM2 protein complex. Activated p53 are further modified by various modifications, including phosphorylation, acetylation, methylation, etc. Modified p53 triggers cells to undergo cell-cycle arrest, either to proliferation after successful DNA repair or senescence without repair. Moreover, p53 can lead to apoptosis when the chromatin damage is too severe.
Accumulated p53 is further modified to its active form with multiple post-translational modifications. Overlapping with the stabilization step, ATM, ATR and DNA-PK kinases phosphorylate Ser15 of p53 [134-137]. Thr18 of p53 is phosphorylated by casein kinase 1 (CK1) [138]. The checkpoint kinase 1/2 (Chk1/2), which are the substrates of ATM and ATR, phosphorylate Ser20 of p53 [139]. These key phosphorylation sites within transactivation domain are crucial for the dissociation of p53 from MDM2/MDM4, and guide a range of modifications in an intricate but ordered way. The covalent modifications tailor p53 with enhanced activities in DNA binding ability, tetramerization and gene activation. For example, the HAT p300/CBP, which is recruited to p53 after S15/37 phosphorylation, acetylates lysine residues 373 and 382 at the C-terminal regulatory domain to increase the association of p53 to the p21 promoter region [140]. In addition, acetylation of K120 and K164 within DNA-binding domain by HATs MOF/Tip60 and p300/CBP, respectively, promotes specific binding of p53 to the p21 and PUMA promoters [141,142]. Further, methylation of p53 at C-terminal residue K372 contributes to the p21 gene activation by increasing the affinity between p53 and the p21 promoter [143]. After these activation steps, p53 binds to its target genes with specific recognition elements and recruits various cofactors to fine-tune transcription, which may lead to diverse downstream events such as cell-cycle arrest, apoptosis and DNA repair (Figure 2) [144].
Interestingly, p53 can recognize DNA fragments via non-specific manner to participate in DNA repair process [145]. Moreover, p53 itself may translocate to cytoplasm and facilitate transcription-independent apoptosis [146]. Since the different roles of p53 are important in deciding different cellular outcomes, it will be discussed with more details in the following paragraphs.
p53-mediated pathways with different cell fates
As a cellular gatekeeper, p53 is always accountable for answering the question about “to be or not to be”, i.e., the choice between to live or to die after DNA damages [124].
Cell-cycle arrest: Upon activation by DNA damages, p53 serves as a transcriptional factor to regulate the expression of genes involved in the cell-cycle arrest. The cell cycle includes four distinct stages, the G1, S, G2 and M phases. Multiple checkpoints are involved in the whole cell cycle to guarantee the accuracy of the cell division [147]. The G1/S checkpoint blocks DNA synthesis if DNA damage is detected. If there is no DNA damage present, several cyclin/cdk complexes sequentially work to phosphorylate the tumor suppressor pRb, leading to the dissociation of transcription factor E2F from pRb and the activation of genes important for DNA replication initiation [148] p53 controls the G1/S cell cycle arrest by activating p21, which inhibits cyclin E/cdk2 and cyclin A/cdk2 complexes [149].
Another important DNA damage checkpoint occurs at the G2/M phase transition. The cyclin B/cdc2 complex is the main target for the G2/M phase checkpoint. p53 upregulates p21 and GADD45, which in turn induce the dissociation of cdc2 from cyclin B [150,151]. 14- 3-3σ, another p53 target, promotes sequestration of cdc2 complex in cytoplasm [152,153]. Meanwhile, p53 can directly repress Cdc25C, which dephosphorylates the cdc2 complex for activation [154]. In sum, p53 maintains cell cycle arrest by regulating its target genes related to the DNA damage checkpoints at the G1/S and G2/M transitions. On the other hand, p53-deficient cells have failures in pausing at the S phase and mitosis checkpoints [155-158].
DNA damage repair: Since cell-cycle checkpoints always postpone progression of the cell cycle under DNA damage conditions, it is considered as a surveillance mechanism to allow enough time for DNA repair. Although p53 is not a primary sensor of DNA damage stresses, it functions as a critical effector through transcriptional regulation or association with certain DNA repair factors. At the p53-depenent transcription aspect, xeroderma pigmentosum group C (XPC) is a p53 target gene and the protein senses pyrimidine dimers and recruits TFIIH complex to the DNA damage sites during NER [159-161]. As mentioned before, p53 transactivates WIP1, which can remove DNA damage marker γH2AX to restore the chromatin to pre-damage status [55]. In addition, p53 is able to directly interact with some proteins involved in DNA repair. For example, p53 dimethylated at K370 and K382 after DNA damage has higher affinity to 53BP1, which may enhance the involvement of p53 during the repair process [162]. More strikingly in HR repair, p53 can inhibit heteroduplex formation mediated by RAD51 due to their direct association [163-165]. p53 is found to recognize several DNA structures including single-strand DNA, DSB ends, DNA with holiday junctions, DNA duplex with insertion-deletion-lesion mismatches, DNA with single-stranded gap, and triple-stranded DNA [145]. These DNA structures usually appear as the intermediates of DNA damage and repair, and associate with p53 via non-specific sequence binding manners [145]. Additionally, p53 is reported to facilitate reannealing and end-joining at DNA damage sites during NHEJ [166,167]. However, the direct roles of p53 in these damaged DNA structures are yet to be explored.
Apoptosis: When DNA repair machinery is helpless to recover from extensive DNA damages, p53 triggers apoptosis via transcriptiondependent or –independent pathways, to terminate those cells that may do damage to the whole organism. On one hand, p53 is known to induce expression of pro-apoptotic genes, such as PUMA (p53 upregulated modulator of apoptosis), BAX (Bcl2-associated protein X), and BAK (Bcl-2 antagonist/killer) [168-170]. PUMA and some other factors lead to the release of BAX and BAK from anti-apoptotic proteins. Activated BAX and BAK then increase the permeability of mitochondria membrane, and induce cytochrome c release and apoptosis [171,172]. On the other hand, p53 itself can be targeted to and accumulate in mitochondria for apoptosis [173,174]. The p53P72 polymorphism has been shown to have greater ability in mitochondria localization and apoptosis induction than the p53P72 allele [175]. Although there is no mitochondria-targeting sequence identified in p53, some references reveal that Tid1 (tumorous imaginal discs 1) or OKL38 (ovary, kidney and liver protein 38) may help to regulate p53 mitochondrial translocation [176,177].
In short, p53 functions as a “cellular rheostat” [115], which can receive and gauge the upstream signals, transfer them to downstream effectors with more specific cellular functions leading cells to their unique fates and destinations (Figure 2).
The DNA damage response signaling cascade is initiated by detection of DNA breakage sites (Figure 3). The sensors pass the damage messages to transducers that are mainly kinases, which pass on the signals to modulate chromatin structures by many factors, such as histone modification enzymes, chromatin remodelers, histone chaperones. The altered chromatin structure facilitates and coordinates many events critical for repair of the damaged chromatin. Chromatin binding proteins, such as the PWWP domain containing proteins, are effectors recruited to the DNA damage sites to facilitate the repairing process. However, these proteins can further crosstalk with the chromatin modifiers via feedback loops to reinforce the function of the DNA damage repair machineries. Furthermore, the p53 tumor suppressor can regulate the expression of downstream target genes to determine the cellular outcomes after DNA damage, including cell cycle arrest, senescence or apoptosis. It is notable that cytoplasmic p53 plays a direct role in apoptosis via a mitochondrial mediated pathway. In sum, chromatin maybe viewed to play a pivotal role in organizing the entire DNA damage response and facilitating the cellular fate determination after DNA damages.
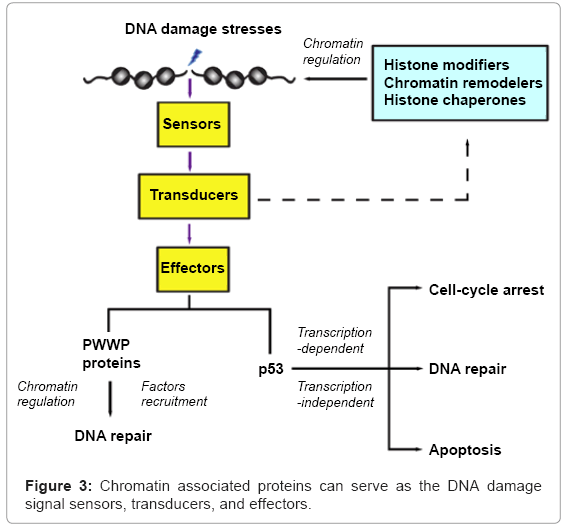
Figure 3: Chromatin associated proteins can serve as the DNA damage signal sensors, transducers, and effectors.
The author thanks members of the Wang lab as well as the Center for Eukaryotic Gene Regulation for helpful discussions. We thank Dr. Kuangyu Yen for critical reading of this review. Jing Hu is partially supported by the BMMB graduate program. Research in the Wang lab is supported by NIH R01 CA136856 to Y.W.