Journal of Glycobiology
Open Access
ISSN: 2168-958X
ISSN: 2168-958X
Research Article - (2014) Volume 3, Issue 1
Keywords: N-Glycan; Endo-β-N-acetylglucosaminidase; Gene therapy; In vivo gene transfer; N-acetylglucosamin; B16 Melanoma; Immune response
Cell surface glycans are carbohydrate structures composed of glycoproteins and glycolipids, and are important for various biological processes including development, cellular differentiation, and migration, all of which are controlled by a signal transduction network [1]. Foreign glycans are often recognized by the immune system as antigens and allow for the prevention of bacterial or viral infection [2]. The synthesis of the carbohydrate chains of glycans is catalyzed by glycosyltransferases and glycosidases. Since one enzyme per glycosidic linkage is required for the synthesis of oligosaccharides, more than 100 types of glycosyltransferases appear to exist in mammalian cells [3,4]. Many glycosidases that catalyze the hydrolysis of the glycosidic linkage to release smaller sugars have been found in all organisms [5]. Glycosidases are present as intracellular and extracellular enzymes with roles in nutrient acquisition [6].
In higher organisms, glycosidases are localized in the endoplasmic reticulum (ER) and Golgi apparatus, where processing of N-linked glycoproteins occurs, and in them lysosome, where they are involved in the degradation of carbohydrate chains [7,8].
Both glycosyltransferases and glycosidases are necessary to maintain the proper structure of glycans associated with glycoproteins and glycolipids [9-13]; furthermore, they are now recognized as powerful and valuable tools to analyze the biological function of glycans [14-16]. To assess the function of the glycan αGal epitope (Gal-α1-3Gal-R) expressed on almost all mammalian cells, except for human and Old World monkeys [17], we first intended to use endo-β-galactosidase C (EndoGalC), a glycosidase isolated from Clostridium perfringens [16,18]. This enzyme removes the αGal epitope by cleaving the Galβ1- 4GlcNAc linkage in the Galα1-3Galβ1-4GlcNAc sequence [19]. A previous report indicated that recombinant NIH3T3 (mouse derived fibroblast) cells transfected with an EndoGalC expression vector lacked αGal epitope expression on their cell surface, and exhibited 1.8-fold higher proliferation activity than untransfected parental cells [16]. This accelerated cellular proliferation in the recombinant NIH3T3 cells was caused by ligand-independent activation of the transforming growth factor β (TGFβ) receptor (TβR). To test whether the recombinant NIH3T3 cells can form solid tumors in vivo like other cancer cells, we subcutaneously transplanted them into nude mice.
Surprisingly, the NIH3T3 cells failed to form tumors, although the concomitantly transplanted untransfected wild-type cells did form tumors (unpublished data). Notably, Huflejt et al. [20] reported that more than 96% of humans have serum antibodies capable of binding to the GlcNAc residue. In light of this evidence, we supposed that the immune system of the nude mice reacts against GlcNAc residues exposed on the cell surface after EndoGalC digestion, leading to growth suppression of the grafted cells. With respect to this supposition, the GlcNAc residue can be recognized as an in vivo ‘anti-tumor antigen’ for cancer vaccination. Cancer vaccination is one of the immunotherapeutic strategies against tumors [21,22]. It can be performed by introduction of cancer-associated or -specific antigens into individuals or of DNA itself encoding these proteins or genetically modified tumor cells engineered to express those molecules. The cancer-related molecules expressed in the individuals can be recognized by immunomodulatory cells such as dendritic cells, which in turn evoke specific immune response against tumor cells [23]. To induce autologousimmune response, adeno- or retrovirus vectors have been employed for transfer of antitumor molecules such as cytokine costimulatory factor, granulocyte macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), CD40, CD80 as phase II or III clinical trial [22]. Notably, among these trials, a gene encoding mouse α1,3 galactosyltransferase (α1,3GalT) was used to induce αGal epitope expression in tumor cell surface [24] (ClinicalTrials.gov Identifier: NCT00105053). This means that specific modification of cell surface glycan can lead to successful elimination of tumor cells in vivo. To evaluate our hypothesis that the GlcNAc residue exposed after digestion with glycosidase can act as an in vivo ‘anti-tumor antigen’, we decided to use another glycosidase, human endo-β-N-acetylglucosaminidase (hENGase) [25], which cleaves the amide bond between the proximal GlcNAc residues at the side chain of an asparagine residue on N-glycans. This cleavage results in the exposure of GlcNAc residues on the cell surface (Figure 1B). We cloned hENGase cDNA and constructed an hENGase expression vector. The recombinant B16 melanoma cells overexpressing hENGase were then produced, and their properties were assessed with respect to the level of exposure of GlcNAc residues, the tumor-forming ability upon grafting, and the possibility of using an hENGase expression vector as a tool for cancer vaccination.
Biological activity of hENGase
To assess the biological activity of hENGase, a 9.2-kb fragment containing ChENGn (hENGase expression vector; Figure 1A) was liposomally introduced into the genome of B16 melanoma cells. After selection with G418, we obtained 6 stable recombinants, termed B6- hENG-1 to -6. Two of these recombinant lines were first subjected to western blot analysis to detect ENGase protein using an anti-ENGase antibody. As expected, an increased intensity of an 84-kDa protein, corresponding to the hENGase protein expressed from pChENGn (hENGase expression vector containing neo gene), was visible in the hENGase-expressing recombinants (B6-hENG-1 and -2) (Figure 2A).
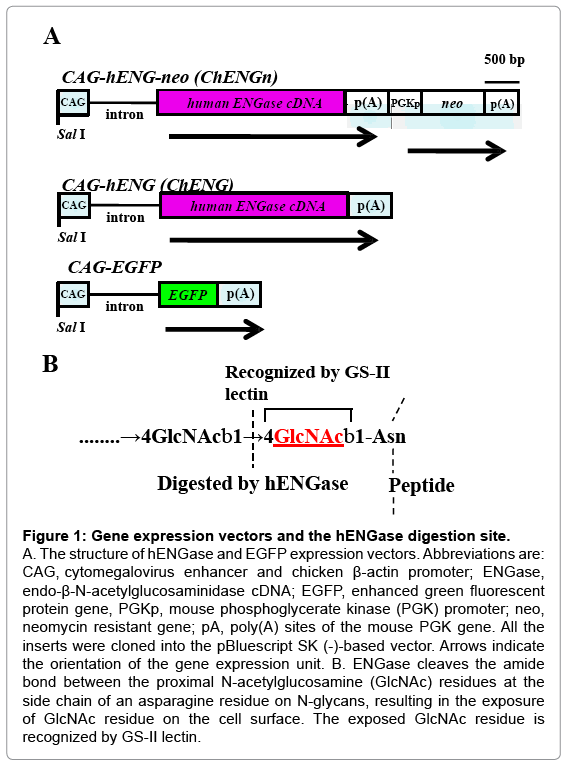
Figure 1: Gene expression vectors and the hENGase digestion site.
A. The structure of hENGase and EGFP expression vectors. Abbreviations are: CAG, cytomegalovirus enhancer and chicken β-actin promoter; ENGase, endo-β-N-acetylglucosaminidase cDNA; EGFP, enhanced green fluorescent protein gene, PGKp, mouse phosphoglycerate kinase (PGK) promoter; neo, neomycin resistant gene; pA, poly(A) sites of the mouse PGK gene. All the inserts were cloned into the pBluescript SK (-)-based vector. Arrows indicate the orientation of the gene expression unit. B. ENGase cleaves the amide bond between the proximal N-acetylglucosamine (GlcNAc) residues at the side chain of an asparagine residue on N-glycans, resulting in the exposure of GlcNAc residue on the cell surface. The exposed GlcNAc residue is recognized by GS-II lectin.
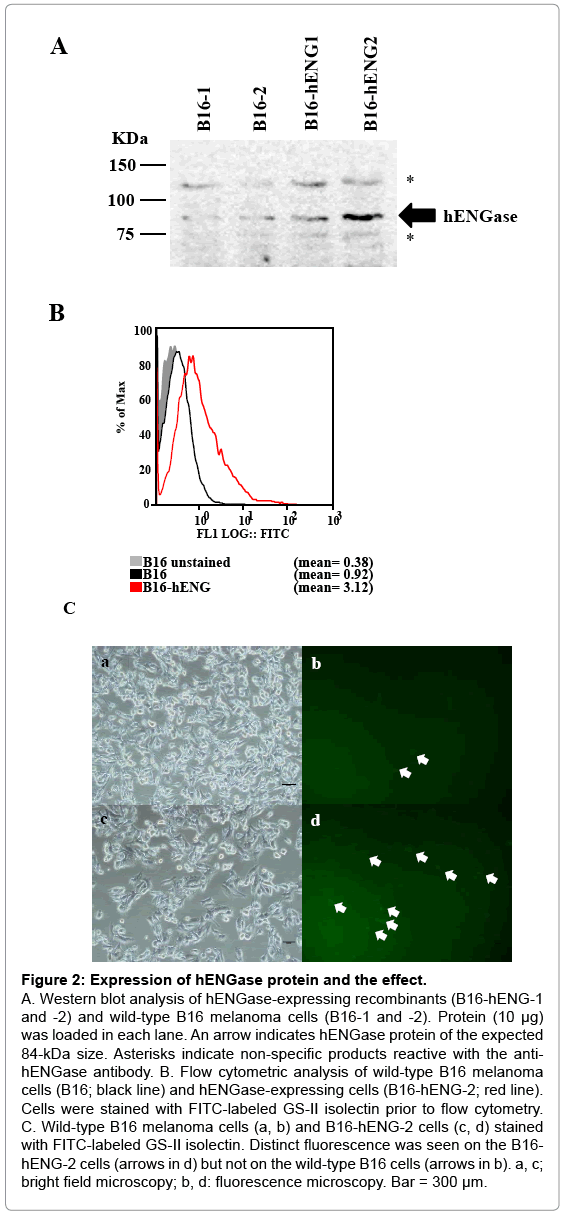
Figure 2: Expression of hENGase protein and the effect.
A. Western blot analysis of hENGase-expressing recombinants (B16-hENG-1 and -2) and wild-type B16 melanoma cells (B16-1 and -2). Protein (10 μg) was loaded in each lane. An arrow indicates hENGase protein of the expected 84-kDa size. Asterisks indicate non-specific products reactive with the antihENGase antibody. B. Flow cytometric analysis of wild-type B16 melanoma cells (B16; black line) and hENGase-expressing cells (B16-hENG-2; red line). Cells were stained with FITC-labeled GS-II isolectin prior to flow cytometry. C. Wild-type B16 melanoma cells (a, b) and B16-hENG-2 cells (c, d) stained with FITC-labeled GS-II isolectin. Distinct fluorescence was seen on the B16- hENG-2 cells (arrows in d) but not on the wild-type B16 cells (arrows in b). a, c; bright field microscopy; b, d: fluorescence microscopy. Bar = 300 μm.
Also, in the control samples (B6-1 and -2), a band or an 84-kDa protein that appears to correspond to the endogenous mouse ENGase was detected by the antibody. This detection is due to the high degree of similarity between the amino acid sequences of mice and human ENGase proteins (72%) [25]. Other bands corresponding to 120 and 74 kDa (shown by asterisks in Figure 2A) appear to be non-specific products. Of the 2 hENGase-expressing recombinants tested, the B6- hENG-2 line that expressed hENGase relatively strongly was selected for further study. We next evaluated the biological activity of hENGase by flow cytometric analysis. As previously shown in Figure 1B, cells producing hENGase should expose GlcNAc residues on their surface. The exposed GlcNAc residue is easily recognized by a specific lectin GS-II [26]. Therefore, the wild-type B6 melanoma and B6-hENG-2 cells were subjected to staining with FITC-labeled GS-II prior to flow cytometric analysis. The B6-hENG-2 recombinants had approximately 3-fold more cell-surface GlcNAc residues than their parental B6 cells (Figure 2B). Microscopic observation of these lectin-stained cells confirmed that the surfaces of B6-hENG-2 recombinant cells were more intensely stained than the surfaces of the control B6 cells (Figure 2C).
Evaluation of the tumor-forming ability of hENGaseexpressing recombinant cells
As previously mentioned, murine cells exhibiting exposed GlcNAc residues on their surfaces cannot survive in vivo when transplanted into host mice (unpublished data). To confirm this possibility, we subcutaneously transplanted hENGase-expressing recombinant B16 cells (B6-hENG-2) into the dorsal region of C57BL/6 mice. The recipient mice were sacrificed to evaluate tumor growth 14 d after transplantation. Surprisingly, the size of tumors derived from the B16-hENG-2 cells remained quite small, compared to those from the wild-type B6 melanoma (Figure 3A); the tumor weight of the B16- hENG-2 cells was about 1/5–1/3 that of the wild-type cells (Figure 3B). Aside from the difference in size between these 2 types of tumors, no appreciable macroscopic differences were observed between them. The hematoxylin-eosin -stained sections of the dissected tumors revealed the presence of extensive cell death (as exemplified by pyknotic nuclei, arrowheads in Figure 3C-b) and invasion by macrophages (arrows in Figure 3C-b) in the samples derived from B16-hENG-2. These results suggest the involvement of an immune reaction exclusively in the hENG-expressing tumor cells. In contrast, the wild-type tumors did not exhibit such events (Figure 3C-a). These results strongly suggest that GlcNAc residues exposed on the surface of the ENGaseexpressing recombinant cells are the target recognized by the immune system. In conclusion, the exposed GlcNAc residues on the surface of recombinants might attenuate the tumor-forming ability of the cells.
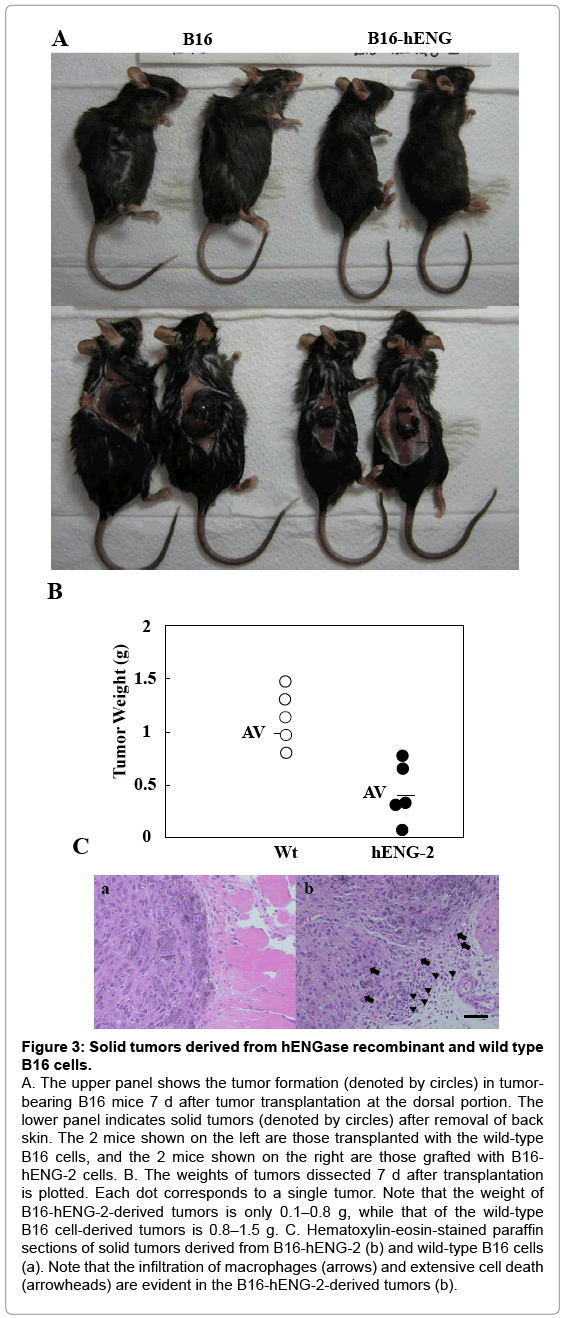
Figure 3: Solid tumors derived from hENGase recombinant and wild type B16 cells.
A. The upper panel shows the tumor formation (denoted by circles) in tumorbearing B16 mice 7 d after tumor transplantation at the dorsal portion. The lower panel indicates solid tumors (denoted by circles) after removal of back skin. The 2 mice shown on the left are those transplanted with the wild-type B16 cells, and the 2 mice shown on the right are those grafted with B16- hENG-2 cells. B. The weights of tumors dissected 7 d after transplantation is plotted. Each dot corresponds to a single tumor. Note that the weight of B16-hENG-2-derived tumors is only 0.1–0.8 g, while that of the wild-type B16 cell-derived tumors is 0.8–1.5 g. C. Hematoxylin-eosin-stained paraffin sections of solid tumors derived from B16-hENG-2 (b) and wild-type B16 cells (a). Note that the infiltration of macrophages (arrows) and extensive cell death (arrowheads) are evident in the B16-hENG-2-derived tumors (b).
Effect of normal mouse serum on the growth of hENGaseexpressing recombinant cells
Growth retardation of the hENGase-expressing B16 recombinant cells suggests the involvement of an immune response in vivo. We theorized that one of these responses could involve the host anti- GlcNAc antibody. To assess this possibility, wild-type B16 melanoma and B16-hENG-2 cells were cultured in medium containing 10% (v/v) non-heat-inactivated normal mouse serum for 1 d. This serum is thought to contain many types of antibodies, including the anti-GlcNAc antibody. When cells were assessed for cell death using a LIVE/DEAD cell viability assay kit, extensive cell death (denoted by the presence of red fluorescent cells) was observed in the B16-hENG-2 sample cultured with mouse serum-containing medium (Figure 4A-a). In contrast, only a few cells were found to be dead when wild-type B16 melanoma cells were treated with mouse serum (Figure 4A-b). No appreciable cell death was noted when these 2 types of cells were cultured in the presence of heat-inactivated fetal bovine serum (FBS) (Figure 4A-c,d). Notably, the number of SYTOX-green (Invitrogen, Carlsbad, CA, USA)-stained cells was greatly reduced in the B16-hENG-2 sample treated with mouse serum, compared to other treatment group (a versus b, d in Figure 4A). This result suggests that extensive cell death had occurred within the 24-h incubation, and that these dead cells might have been removed upon medium change prior to being subjected to cell staining. Furthermore, proliferation of B16-hENG-2 cells cultured in the presence of mouse serum for over 7 d was greatly suppressed, which was in contrast with that of those cultured in FBS-containing medium (data not shown). These results suggest that components (probably immunoglobulin; Ig) included in the mouse serum are deleterious to the hENGase-expressing B16 recombinants, but not to the wild-type B16 cells. To assess the binding of mouse serum Ig to the cell surface of hENGase-expressing B16 melanoma cells, flow cytometric analysis was performed. Wild-type B16 melanoma and B16-hENG-2 cells were incubated in a solution containing 10% of non-heat-inactivated mouse serum for 1 h at 4°C, and cells were then washed with a solution containing phycoerythrin (PE)-conjugated anti-mouse IgM/G for 1 h at 4°C. The B16-hENG-2 cells treated with mouse serum exhibited an approximately 2.6-fold higher degree of binding activity with mouse Ig than the wild-type B16 melanoma cells (Figure 4B). This result strongly suggests that the GlcNAc residue exposed after hENGase expression can be recognized by serum Ig.
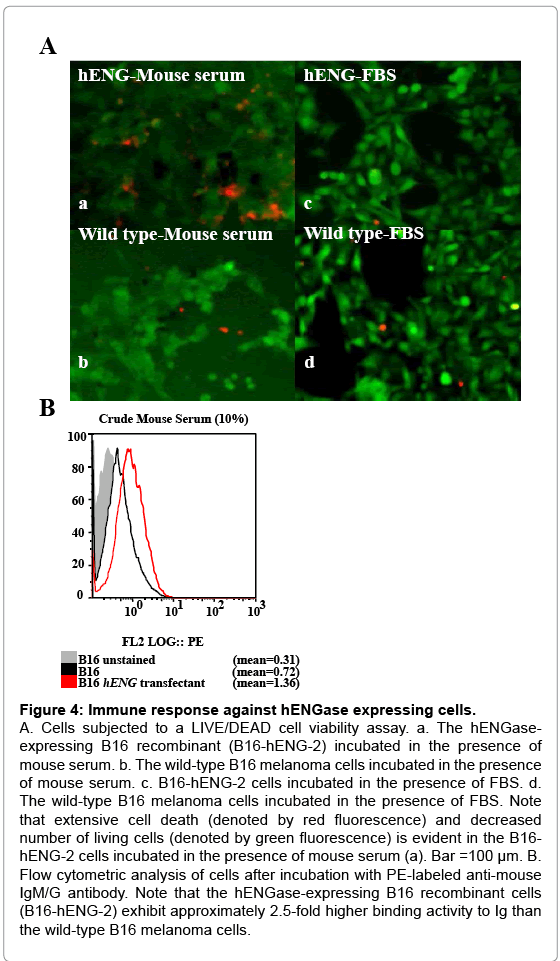
Figure 4: Immune response against hENGase expressing cells.
A. Cells subjected to a LIVE/DEAD cell viability assay. a. The hENGaseexpressing B16 recombinant (B16-hENG-2) incubated in the presence of mouse serum. b. The wild-type B16 melanoma cells incubated in the presence of mouse serum. c. B16-hENG-2 cells incubated in the presence of FBS. d. The wild-type B16 melanoma cells incubated in the presence of FBS. Note that extensive cell death (denoted by red fluorescence) and decreased number of living cells (denoted by green fluorescence) is evident in the B16- hENG-2 cells incubated in the presence of mouse serum (a). Bar =100 μm. B. Flow cytometric analysis of cells after incubation with PE-labeled anti-mouse IgM/G antibody. Note that the hENGase-expressing B16 recombinant cells (B16-hENG-2) exhibit approximately 2.5-fold higher binding activity to Ig than the wild-type B16 melanoma cells.
Use of the hENGase expression vector for cancer vaccination
Our present data suggest that the GlcNAc residues exposed after hENGase production can elicit an immune response, leading to elimination of the hENGase-expressing cells. These data led us to suppose that a hENGase expression vector might be used for cancer vaccination, which is a gene therapy method. In general, cancer vaccination is performed through the modification of cell surface components by expression of a transgene and subsequent elimination of the transgene-expressing cells by the immune response. To test the supposition, we employed an in vivo gene delivery system based on plasmid DNA injection into a target tumor and subsequent in vivo electroporation using tweezer-shaped electrodes. Generally, retrovirus or adenovirus vectors have been used for eliciting cancer vaccination [25-28] which requires the preparation of new vector plasmids and packaging cells. However, our present system does not require such components; therefore, it is simpler than the current methods. B16 melanoma cells were subcutaneously transplanted into the dorsal region of C57BL/6 mice. Seven days after transplantation, an hENGase expression vector (pChENG) was introduced by in vivo electroporation, as shown schematically in Supplementary Figure 1. The pCAG-EGFP vector was also injected as a control. Two days after gene delivery, the DNA-injected tumors were dissected to assess the production of EGFP or hENGase. Tumors were mechanically cut into pieces, digested into single cells, treated with trypsin, and stained with FITC-labeled GS-II lectin. Subsequent flow cytometric analysis revealed the percentage of cells producing either EGFP or FITC fluorescence ranged from 40 - 60% (Figure 5A). This result implies a high degree of efficiency for gene delivery in this system. Seven days after gene transfer, the tumor-bearing mice were sacrificed to assess tumor regression after gene delivery of pChENG or the control plasmid, pCAG-EGFP. The size of the tumors injected with pChENG were smaller compared to the control tumors (Figure 5B and 5C). The average weight of the tumors injected with pChENG was less than 1/2 of those injected with pCAG-EGFP (Figure 5D). These results strongly suggest that a hENGase expression vector can be used for cancer vaccination.
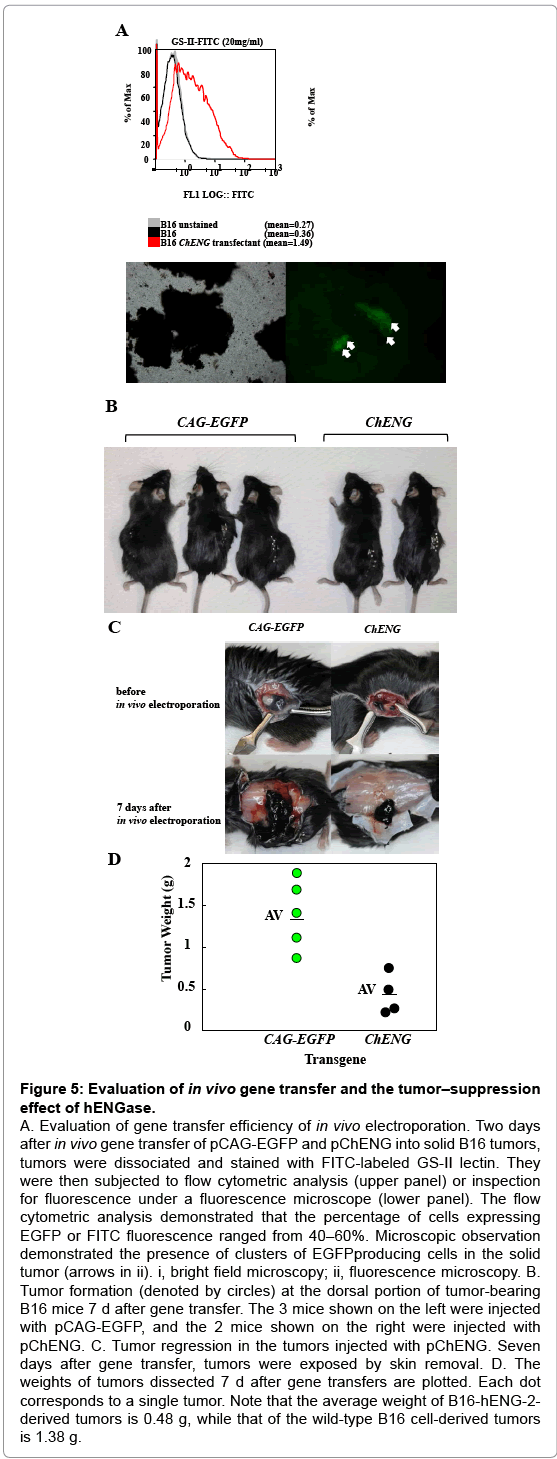
Figure 5: Evaluation of in vivo gene transfer and the tumor–suppression effect of hENGase.
A. Evaluation of gene transfer efficiency of in vivo electroporation. Two days after in vivo gene transfer of pCAG-EGFP and pChENG into solid B16 tumors, tumors were dissociated and stained with FITC-labeled GS-II lectin. They were then subjected to flow cytometric analysis (upper panel) or inspection for fluorescence under a fluorescence microscope (lower panel). The flow cytometric analysis demonstrated that the percentage of cells expressing EGFP or FITC fluorescence ranged from 40–60%. Microscopic observation demonstrated the presence of clusters of EGFPproducing cells in the solid tumor (arrows in ii). i, bright field microscopy; ii, fluorescence microscopy. B. Tumor formation (denoted by circles) at the dorsal portion of tumor-bearing B16 mice 7 d after gene transfer. The 3 mice shown on the left were injected with pCAG-EGFP, and the 2 mice shown on the right were injected with pChENG. C. Tumor regression in the tumors injected with pChENG. Seven days after gene transfer, tumors were exposed by skin removal. D. The weights of tumors dissected 7 d after gene transfers are plotted. Each dot corresponds to a single tumor. Note that the average weight of B16-hENG-2- derived tumors is 0.48 g, while that of the wild-type B16 cell-derived tumors is 1.38 g.
We aimed to explore the possibility that a vector encoding an enzyme that digests cell surface carbohydrates could be a useful tool for developing cancer vaccination strategies. This possibility is based on our previous finding that cells transfected with an EndoGalC expression vector lost the αGal epitope from their surfaces and had exposed GlcNAc residues, which lead to host immune reactions upon grafting cells into a host (unpublished data). To examine the immunogenicity of cell surface carbohydrates in more detail, we employed B16 melanoma cells; they are easy to detect because of active melanization, and they are highly malignant. The latter property is particularly beneficial for evaluating the effects of a carbohydrate digestive enzyme, because visible solid tumors, that are appropriate in size for performing gene delivery experiments, are generated 7 d after grafting. Interestingly, αGal epitope were not found on B16 melanoma, like cells derived from Old Word monkeys and humans though they were derived from C57BL/6 mouse [29]. Therefore we decided to use hENGase capable of digesting carbohydrate chains at the basal portion of N-glycan and make the GlcNAc residue be exposed on cell surface.
Consistent with our hypothesis, gene delivery of a hENGase expression vector to solid B16 melanoma tumors resulted in efficient inhibition of tumor formation. For example, we observed cell death when hENGase-expressing B16 melanoma cells were cultured in the presence of non-heat-inactivated mouse serum (Figure 4A). Furthermore, we clarified that this phenomenon is caused by an anti-GlcNAc residue antibody from normal serum (Figure 4B). Thus, it is likely conceivable that cancer cell ablation after gene delivery of hENGase construct is caused as a result of general immune response, in which anti-GlcNAc antibody present in serum binds to the terminal GlcNAc residues exposed after hENGase-mediated digestion of carbohydrate chains, and subsequently complement-mediated cytotoxicity occurs, as previously reviewed by Tanemura et al. [30]. In agreement with our findings, Huflejt et al. [20] reported that more than 96% (102/106) of human subjects had serum antibodies that bind to GlcNAc residues.
Why does the anti-GlcNAc antibody exist in mammals including mice? Cooper et al. demonstrated that anti-GlcNAc antibody is generated by natural infection with group A streptococci, bacteria present in almost all of the mammals including humans, and thus is defined as the antibody recognizing a dominant epitope of the group a streptococcal cell wall polysaccharide [31]. Notably, S. pyrogenes, one of the group A streptococci that belong to normal bacterial flora, is known to cause adenoiditis and sometimes blood poisoning septicemia. Remarkably, the anti-GlcNAc antibody that is known to bind to cytokeratin peptide and myosin in myocardium is also considered to elicit acute myocarditis upon infection with group A streptococci [32]. LaTemple et al. [33] demonstrated that the α1,3- galactosyl transferase (αGalT) gene can be a useful tool for cancer vaccination. The αGalT enzyme creates an αGal epitope, a xenoantigen, at the end of N-glycans. In humans and Old Word monkeys, this αGal epitope is absent as a result of mutations in the αGalT gene [34]. If αGal epitope-expressing cells, such as porcine cells, are grafted into human tissues, hyper acute rejection occurs [35]. This rejection is due to an immune reaction caused by the anti-αGal antibody present in human sera. Galili et al. [36] produced genetically modified human cells capable of overexpressing the αGal epitope by introduction of an αGalT expression vector, and found that these engineered cells were rapidly eliminated under cultivation in the presence of human sera. Since the first report by Galili et al. [36] appeared, many studies have focused on glycotransferase as a promising tool for cancer vaccination [30]. However, the possibility glycosidase as a useful tool for cancer vaccination has been widely overlooked. In this study, we employed an in vivo electroporation approach to perform efficient gene delivery to a target tumor and found that about 50–60% of tumor cells were successfully transfected. Aside from the high degree of gene transfer efficiency, the voltage of the pulse used in this study was only 50V, which is 1/5–1/3 of the voltage generally used for in vitro gene transfer [37].This approach is of practical value because of its low invasiveness. In support of our approach, Niwa et al. [38] demonstrated the feasibility of in vivo electroporation in cancer vaccination in their review.
Our present findings demonstrated that introduction of a hENGase expression vector can be used for gene therapy against malignant tumors. If an efficient and cancer-specific delivery system is developed, our method could be a beneficial component of cancer vaccination. In addition to tumor cell elimination, this ENGase-based gene transfer system could be a useful tool for specific cell ablation studies in embryos and adults. In summary, we have demonstrated that combinational use of a hENGase expression vector and in vivo electroporation is effective as a powerful cancer vaccination tool for the attenuation of malignant tumor growth.
Ethics statement
This study was approved by the Animal Care and Use Committee of the National Institute of Agrobiological Sciences (NIAS) (Permission number: H18-040-1) and carried out according to the Guide for the Care and Use of Laboratory Animals at the NIAS. All efforts were made to minimize the number of animals used and their suffering.
Vector construction
A vector for overexpressing hENGase cDNA was constructed by inserting the gene into the mammalian expression vector pCAGGS [39]. The hENGase cDNA was first amplified by RT-PCR with the primers hENGase-S 5’-ggaattcacagtcatggaggccgcggcggtgac-3’ and hENGase-R 5’-ggaattcgctcatgcaggggctgaata-3’ using 293T cell-derived mRNA [40] and KOD polymerase (TOYOBO Co., Tokyo, Japan). The resulting 2.6-kb amplified fragment was then subcloned into the EcoRI site of pBluescript II SK (+) (Stratagene, La Jola, CA, USA), and the fidelity of the insert was confirmed by sequencing. The hENGase cDNA was next subcloned into the EcoRI site of the third exon of the rabbit β-globin gene in pCAGGS. The resulting plasmid, termed pChENG (Figure 1A), has hENGase cDNA in the sense orientation. The PGK-neo-p (A) cassette, obtained after digestion of pKJ2X+ [37] with EcoRI and XhoI, was introduced into the BamHI site of pChENG by blunt-end ligation. The resulting plasmid, termed pChENGn (Figure 1A), was subjected to sequencing to check for proper insertion. A 9.2 kb fragment containing ChENGn was isolated by digestion of pChENGn with SalI and subsequent size-fractionation in a 0.8% agarose gel (#01157-66; Nacalai Tesque Co., Kyoto, Japan), prior to gene transfer.
Cell culture and transfection
B16 mouse derived melanoma cells [41] were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich Co. Ltd., St. Louis, MO, USA) supplemented with 10% FBS, 50 units of penicillin, and 50 μg/mL of streptomycin at 37°C in an atmosphere of 5% CO2 in air. For transfection, B16 cells (2×105) were first seeded onto a 60-mm dish (#3002; Becton Dickinson, Franklin Lakes, NJ, USA). The next day, the 9.2-kb DNA fragment containing ChENGn (5 μg) encapsulated by FuGENE HD reagent (Roche Diagnostics, Mannheim, Germany) was added to the cell culture. The cells were passaged onto 2100-mm dishes (#3003; Becton Dickinson) 24 h after transfection. One day after the passage, the cells were selected by culturing in a medium containing 500 μg/mL G418 (#10131-027; Invitrogen, Carlsbad, CA, USA) for 7 d. The surviving colonies were picked up, transferred to a 24-well plate, and then propagated in a fresh 60-mm dish until confluence. The stable B16 transfectant carrying pChENGn was termed B16-hENG.
Western blot analysis
Cells were homogenized in TNE buffer [10 mM Tris-HCl (pH 7.8), 150 mM NaCl, 1 mM ethylenediamine tetraacetic acid (EDTA), and 1% (v/v) NP-40] containing complete protease inhibitor (#1697498; Roche Diagnostics), according to the manufacturer’s instructions. These proteins were separated by electrophoresis under reducing conditions on 8% sodium dodecyl sulfate polyacrylamide gel and transferred to nylon membranes (Immobilon-P; Millipore, Bedford, MA, USA). These blots were blocked using 1% (w/v) non-fat dry milk in Trisbuffered saline [TBS; 50 mM Tris-HCl (pH 7.4) and 150 mM NaCl] containing 0.05% (w/v) Tween20 (#28353-85; Nacalai Tesque Co.) (TBST) and then incubated with 1 μg/mL of anti-ENGase antibody (#AB107738; Abcam, Cambridge, UK). After washing with TBST, the blot was incubated with horseradish peroxidase (HRP)-linked anti-rabbit IgG antibody (#7074; Cell Signaling Technology, Danvers, MA, USA) diluted 3000-fold in TBST. The blots were then re-washed with TBST, and the proteins were detected by treating the membranes with ECL Advance western blotting reagent (#RPN2135; Amersham Pharmacia Biotech, Piscataway, NJ, USA) and analyzed with the aid of a FLA3000 image analyzer (Fuji film Co., Tokyo, Japan). The blots were then washed with WB stripping solution (#05364-55; Nacalai Tesque Co.) to remove the antibodies and blocked again in TBST containing 1% non-fat milk for subsequent re-probing.
Flow cytometric analysis of the recombinant cells
Cells were stained with 20 μg/mL of FITC-labeled GS-II lectin (#F-2402-2; EY Laboratories, San Mateo, CA, USA) in PBS(–)/BSA [Dulbecco’s modified phosphate buffered saline without Ca2+ and Mg2+ (pH 7.2); 0.2% (w/v) bovine serum albumin; 0.1% (w/v) sodium azide] on ice for 30 min to detect the GlcNAc residue exposed on the cell surfaces after digestion with hENGase. After incubation, the cells were washed twice with PBS (–)/BSA, resuspended in 0.5 mL of PBS (–)/BSA, and analyzed using flow cytometry (Epics XL-MCL; Beckman Coulter, Fullerton, CA, USA). The mean fluorescence intensity (MFI) was used to quantify the levels of the exposed GlcNAc residues. The data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA). The value of expression was calculated as % [(MFI of the test cells stained with FITC-labeled lectin – MFI of untransfected cells without staining with FITC-labeled lectin)/(MFI of untransfected cells stained with FITC-labeled lectin – MFI of untransfected cells without staining with FITC-labeled lectin)]. Similarly, to examine the possible binding of mouse IgG to the surface of hENGase-expressing cells, B16 cells were incubated in PBS(- ) containing 10% non-heat-inactivated mouse serum for 30 min. Mouse sera were collected from 5 C57BL6/N males (12 weeks old) and immediately frozen at 80°C prior to use. The cells treated with mouse sera were then washed with PBS (-)/BSA and incubated with 20 μg/mL of phycoerythrin (PE)-conjugated anti-mouse IgG (#731743; Beckman Coulter, Fullerton, CA, USA) antibody in PBS (-)/BSA for 30 min on ice. The cells were washed with PBS (-)/BSA before flow cytometry analysis.
Cell viability assay
The cell viability of the hENGase-expressing cells in medium containing non-heat-inactivated mouse serum was examined. Untransfected wild-type or recombinant B16 cells (each 5×104 cells) were seeded onto 24-well plates, and the medium was changed to DMEM containing 10% non-heat-inactivated mouse serum 24 h later. One day after culture, cells were stained by using the LIVE/DEAD cell viability assay kit (#L3224; Life Technologies Co., Grand Island, NY, USA) according to the manufacturer’s instructions.
Cell transplantation
Male C57BL/6 mice (8 weeks old; SLC Co., Shizuoka, Japan) were used as hosts for transplantation experiments of B16 melanoma cells. Untransfected wild-type or recombinant B16 cells (each 5×106 cells suspended in 0.3 mL of serum-free DMEM) were subcutaneously transplanted into the dorsal region of C57BL/6 male mice with a 27-gauge needle (Terumo, Tokyo, Japan). Ten days after transplantation, mice were sacrificed by cervical dislocation or inhalation of CO2 gas, and the emerging tumors were dissected for several analyses, including the measurement of tumor weight and cellular, biological, and histological analyses.
In vivo gene transfer
Direct in vivo gene transfer to a solid tumor generated under the skin was performed according to the method reported by Sato et al. [42] and is schematically shown in Supplementary Figure 1. The tumorbearing C57BL/6 male mice were obtained as described in the Cell transplantation section by injecting wild-type B16 cells. Ten days after tumor cell transplantation, the mice were anesthetized with sodium pentobarbital (Somunopentyl; Kyoritu seiyaku, Tokyo, Japan). For DNA injection, about 5 μL of a solution containing circular pChENGn DNA (0.5 μg/μL) was injected into the central portion of the tumor using a 25 μL Hamilton syringe (Model 702; Reno, NV, USA).
Immediately after DNA injection, the tumor was placed between tweezer-type electrodes and then administered 8 ×50 ms square-wave pulses of 50 V from a pulse generator (#LF101; NEPA GENE Co. Ltd., Chiba, Japan). Similarly, pCAG-EGFP (Figure 1A) (0.5 μg/μL) DNA was also injected as a negative control. Seven days after gene transfer, the tumors were dissected for several analyses including measurement of tumor weight.
Histological analyses of tumors derived from transplanted melanoma cells
Wild type and hENGase recombinant B16 cells were transplanted subcutaneously in the dorsal region as described above. Seven days after transplantation, the mice were sacrificed, and the B16-derived tumors were recovered. These tissues were fixed with paraformaldehyde. After fixation, the tissue block was embedded in paraffin, then cut in a microtome to the desired thickness (approximately 5 μm), and affixed onto the slide. These sections were then deparaffinized and stained with hematoxylin and eosin, and then observed under a microscope.
We would like to thank Dr. Mitsuru Chiba at Hirosaki University for a gift of B16 mouse derived melanoma cells. This study was supported in part by a Grant-in-Aid for Scientific Research (C) (Grant no. 22592050) from the Ministry of Education, Science, Sports, Culture and Technology of Japan.