Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 0, Issue 0
Previous studies have shown that nicotine modulates GABAergic transmission onto dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNc) mainly via presynaptic mechanisms. However, nicotinic modulation of postsynaptic GABAA receptor function in SNc DAergic neurons is unknown. Employing patch-clamp recording technique in single putative DAergic neurons freshly dissociated from rat SNc, we found that functional α4β2-nAChRs were well expressed on majority (77%) of putative SNc DAergic neurons recorded (type ID neurons), while functional α7-nAChRs were only detected in 23% of recorded putative SNc DAergic neurons (type IID neurons). Smoking relevant concentration of nicotine reversibly increased the amplitude of GABAA receptor-mediated wholecell
currents in 63% of type ID neurons expressing α4β2-nAChRs, but not in those type IID neurons expressing α7-nAChRs, which suggests that postsynaptic GABAA receptors on putative SNc DAergic neurons that contain nAChR α4β2 subunits are significantly modulated by nicotine. Interestingly, nicotine at concentrations from 1 nM to 10 μM produced concentration-independent enhancement effects on GABAA receptor-mediated currents. There was no significant influence of pretreatment with either DHβE (a selective α4β2-nAChR antagonist) or 500 nM nicotine over the nicotinic modulation. In addition, 500 nM nicotine did not affect glutamate or glycine-induced current under our experimental conditions. Collectively, our results suggest that nicotine directly boosts the function of postsynaptic
GABAA receptors, which in turn results in hyperpolarization and reduced excitability of putative SNc DAergic neurons through novel α4β2-nAChR-dependent pathways and/or additional postsynaptic mechanisms.
Keywords: Nicotine; Nicotinic acetylcholine receptor; DAergic neuron; GABAA receptors; Substantia nigra pars compacta
ACSF: Artificial Cerebrospinal Fluid; ACh: Acetylcholine; nAChR: Nicotinic Acetylcholine Receptors; DAergic: Dopaminergic; VTA: Ventral Tegmental Area; SNc: Substantia nigra pars compacta; GABAergic: Gamma-Aminobutyric Acid-Ergic; Glu: Glutamate; Gly: Glycine; Bic: Bicuculline; CNS: Central Nervous System; PD: Parkinson’s Disease; PBS: Phosphate-Buffered Saline; TH: Tyrosine Hydroxylase; H-current: Hyperpolarization-activated cation current; RT: Room Temperature.
Dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNc) intermingled with gamma-aminobutyric acid-ergic (GABAergic) neurons play a critical role in regulation of voluntary motor coordination, and selective loss of SNc DAergic neurons results in Parkinson’s disease (PD) [1,2]. Compared to glutamatergic synaptic inputs, SNc DAergic neurons receive significantly more GABAergic inhibitory inputs [3] from the core of nucleus accumbens, striatum, globus palliuds, dorsolateral ventral pallidum, rostromedial tegmental nucleus, as well as substantia nigra pars reticulate [4-11]. These inhibitory inputs are believed to be key elements of nigral microcircuitry [12-14] and represent the key source of inhibitory modulation on DAergic neuron function in the SNc [5,15,16]. Evidence from in vivo single-unit extracellular recordings shows that local pressure application of selective GABAA receptor antagonists, such as bicuculline (Bic) and picrotoxin, results in bust firing of DAergic neurons in rat SNc, suggesting that the activity of SNc DAergic neurons is tonically suppressed by endogenous GABAergic inputs mediated by GABAA receptors [6].
Neuronal nicotinic acetylcholine receptors (nAChRs), members of the superfamily of pentameric ligand-gated ion channels [17,18], are widely distributed throughout the brain. Functional alterations in nAChR-mediated cholinergic pathways are found to be involved in a variety of neurodegenerative disorders, such as PD, Alzheimer’s diseases [19-21], ageing [22,23], and epilepsy [24,25]. A large amount of nAChRs are expressed in mesencephalic DAergic neurons [26-30]. In the SNc, nAChRα2, α4, α5, α6, β2, and β3 subunit mRNAs were detected in most of tested DAergic neurons, α3 and α7 mRNAs were found in about 50% of tested DAergic neurons [31]. Our previous studies, using in situ hybridization, immunohistochemistry, reverse transcriptase polymerase chain reaction (RT-PCR), and electrophysiology, further demonstrated that functional α4β2- and α7- nAChRs are densely expressed on SNc DAergic neurons [32,33]. Bath application of 1 μM nicotine can significantly enhance the inhibitory inputs to the SNc by activating both α7- and non α7-nAChRs at presynaptic terminals in midbrain slices containing the SNc [28]. Moreover, co-localization of α7-nAChRs with GABAA receptors is specifically found at the tips of filopodia extending from dendrites on developing interneurons in the hippocampus [34]. The activation of postsynaptic α7-nAChRs by nicotine or endogenous acetylcholine (ACh) has been reported to impose a reversible inhibition on GABAA receptor function on freshly dissociated chick ciliary ganglion neurons and CA1 interneurons in hippocampal slices [35,36], suggesting that GABAA receptor function can be modulated by postsynaptic α7-nAChRs. Our recent studies using patch clamp recordings in mechanically dissociated SNc DAergic neurons (containing presynaptic GABAergic boutons) found that nicotine not only significantly increases sIPSC frequency, but also dramatically enhances sIPSC amplitude [37], which could not be simply interpreted by presynaptic mechanisms. Our hypothesis is that nicotine may also exert its modulatory effects on postsynaptic GABAA receptor function and increase the amplitude of sIPSCs through some postsynaptic mechanisms.
In the present study, we tested our hypothesis using patch-clamp recordings in single putative SNc DAergic neurons freshly dissociated from rat SN slices. We found that nicotine did modulate somatodendritic GABAA receptor function in complex manners.
Single DAergic neuron dissociation
Wistar rats of either sex, postnatal day 14 (P14)-P21 (averaged 15.9 ± 0.2 days old), were used. All animal procedures were performed in according with the protocol approved by the Institutional Animal Care and Use Committee of Barrow Neurological Institute.
Single DAergic neurons were acutely dissociated from rat SNc as described previously [30,38]. Briefly, after isoflurane anesthesia and decapitation, the brain tissue was removed rapidly and placed in icecold freshly made artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 5 KCl, 24 NaHCO3, 1.3 MgSO4, 1.2 KH2PO4, 2.4 CaCl2, and 10 glucose; pH 7.4; saturated with 95% O2-5% CO2. Two 400-μm midbrain slices containing the SNc were obtained using a vibratome (Vibratome 1000 Plus, St Louis, MO, USA). Then, the slices were incubated in ACSF, continuously bubbled with 95% O2-5% CO2, at room temperature (RT, 22 ± 1°C) for at least 1 hour. Thereafter, the slices were treated with pronase (1 mg/6 ml ACSF) at 31°C for 30-40 min. After enzyme treatment, the SNc was identified according to rat brain atlas [39], then small SNc fragments were punched out from the slices using a well-polished needle with aid of a stereo microscope. Each punched tissue fragment was dissociated using fire-polished micro- Pasteur pipettes in a 35-mm culture dish filled with well-oxygenated freshly made standard extracellular solution containing (in mM): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose and 10 HEPES; pH adjusted to 7.4 with Tris-base. The freshly dissociated cells were going to be ready for recording after they adhered to bottom of the culture dish within 30 min.
Perforated patch-clamp whole-cell recordings
Perforated patch-clamp recordings [30,38,40,41] were employed in this study. The pipette was filled with internal solution containing (in mM): 140 KCl, 4 MgSO4, 0.1 EGTA, and 10 HEPES, pH 7.2 (with KOH), which was freshly supplemented with amphotericin B (200 μg/ml) before experiment. Resistance of the recording pipettes was 3-5 MΩ in the standard extracellular solution. After tight seal (>1 GΩ) formation, about 5-20 min was required for conversion to perforated patch mode, and an access resistance of <60 MΩ was accepted to start experiments. All data were acquired at 10 kHz, filtered at 2 kHz, digitized on-line (Digidata 1322 series A/D board, Axon Instruments), and displayed and stored on a PC computer. Rapid application of drugs was performed using a computer-controlled “U-tube” system [30,40,41]. When ACh was used as a nicotinic agonist, 1 μM atropine was routinely added to exclude possible actions mediated via endogenous muscarinic receptors, and this manipulation is known to have no clear effect on ACh-induced currents [32]. All experiments were performed at room temperature.
Immunocytochemistry
Cell staining was done using a method modified as described previously [42,43] after patch clamp recordings. Briefly, neurons were fixed with 4% paraformaldehyde for 10 min, rinsed three times with phosphate-buffered saline (PBS), and treated with Triton X-100 (0.25%, Sigma-Aldrich) for 30 min as a permeabilizing agent. After rinsing four times with PBS, the neurons were blocked in 5% bovine serum albumin for 30 min, and rinsed for 15 min with PBS. Cells were then incubated overnight at 4°C with anti-tyrosine hydroxylase (TH) primary antibody (AB152; Chemicon International, Temecula, CA, USA) diluted 1:200 in PBS, followed by 2 hours at RT. After three rinses with PBS, a secondary antibody (cy3-conjugated, anti-mouse IgG; Sigma-Aldrich) was applied at RT for 60 min (diluted 1:100). Following another three washes with PBS, neurons were incubated in PBS containing avidin-biotin peroxidase complex (ABC elite kit; Vector Laboratories) for 30 min at RT. The levels of TH were visualized by incubating the cells in distilled water containing 0.05% DAB (3, 3`-Diaminobenzidine tetrahydrochloride; Sigma) plus 0.01% of 30% hydrogen peroxide (Sigma) at RT. The reaction was stopped by washing in distilled water. Cells were examined and photographed with a digital camera on our Olympus inverted microscope (Olympus IX-70, Lake Success, NY, USA).
Chemicals and statistics
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) with exception of RJR-2403, which was purchased from Tocris (Ballwin, MO). Whole cell currents induced by ligands were normalized to the corresponding controls, and then the normalized values were used to do statistical analysis and plot the data. Average data were expressed as mean ± S.E.M (Standard Error of the Mean). Student’s t-test was used to compare two groups of data, and one way ANOVA was used for multiple group comparisons. p<0.05 was considered statistical significance.
Identification of SNc DAergic neurons
Under our present experimental conditions, DAergic neurons were the majority type of neurons dissociated from small SNc fragments, but a small percentage of non-DAergic neurons could not be excluded. Thus, it was important for us to identify DAergic neurons. It is well documented that DAergic neurons are almost two-fold larger in size than GABAergic neurons [44]. In line with these observations, our TH staining results showed that all large neurons are TH (+) (Figure 1D, n=19), while small neurons are TH (-) (Figure 1E, n=6). Thus, we only chose larger neurons for patch-clamp recording. In addition, DAergic neurons were further identified by their electrophysiological characteristics as described previously [30]. Briefly, a neuron was characterized as putative DAergic neuron early in the recording based on: (1) it exhibited slow (1.5 ± 0.1 Hz, from 0.6 to 4.1 Hz, n=64) and regular spontaneous action potentials (Figure 1A), (2) DA (10 μM) hyperpolarized the putative DAergic neurons and inhibited their spontaneous firing (Figure 1B), (3) whole-cell current-clamp recordings revealed hyperpolarizationactivated channels by showing ‘sag’-shaped membrane potential changes (Figure 1Ca) in response to -0.2 or -0.3 nA currents, which was applied as 0.1 nA steps from 0.1 nA, and (4) voltage-clamp recordings showed a hyperpolarization-activated cation current (H-current, Figure 1Cb) induced by hyperpolarizing the neuron from -60 to -120 mV in steps of 10 mV from holding potential of -60 mV. The peak amplitude of H-current at -120 mV was -134.1 ± 16.9 pA (Figure 1Cc, n=11). Several lines of evidence have demonstrated that H-current is present in most (~90%) of midbrain DAergic neurons [45,46], but not in GABAergic neurons [47]. Collectively, we are confident that these electrophysiological, immunostaining, and morphological characteristics of the putative DAergic neurons observed in the present study reliably allowed us to identify the recorded cells as DAergic neurons.
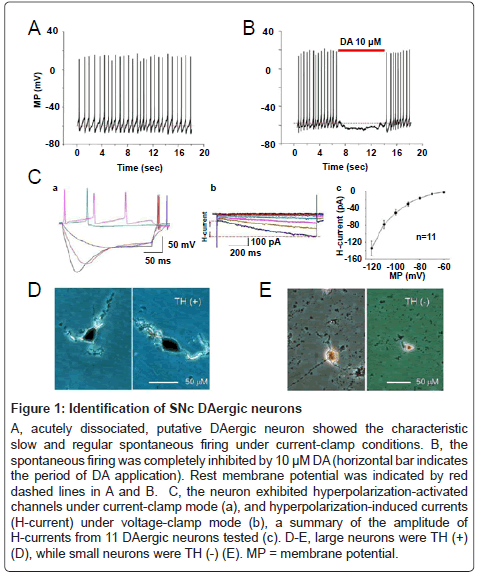
Figure 1: Identification of SNc DAergic neurons
A, acutely dissociated, putative DAergic neuron showed the characteristic slow and regular spontaneous firing under current-clamp conditions. B, the spontaneous firing was completely inhibited by 10 μM DA (horizontal bar indicates the period of DA application). Rest membrane potential was indicated by red dashed lines in A and B. C, the neuron exhibited hyperpolarization-activated channels under current-clamp mode (a), and hyperpolarization-induced currents (H-current) under voltage-clamp mode (b), a summary of the amplitude of H-currents from 11 DAergic neurons tested (c). D-E, large neurons were TH (+) (D), while small neurons were TH (-) (E). MP = membrane potential.
Functional subtypes of nAChRs expressed on SNc DAergic neurons
First, we investigated functional nAChRs naturally expressed on identified SNc DAergic neurons. Under voltage-clamp conditions, nAChR-mediated whole-cell currents were recorded at a holding potential of -60 mV. To identify subunit composition of these nAChRs, ACh (1 mM) or nAChR subunit selective agonists, RJR-2403 (0.1 mM), a selective α4β2-nAChR agonist, or choline (10 mM), a selective α7-nAChR agonist, were rapidly applied to all neurons recorded, respectively. As shown in Figure 2, two types of whole-cell currents were recorded in identified DAergic neurons, suggesting that SNc DAergic neurons mainly express two subtypes of functional nAChRs (ID and IID receptors as previously described in the VTA [30]) based on differential responses to selective nAChR agonists. Type ID receptors were strongly activated by ACh (1 mM, Figure 2A, left) and RJR-2403 (0.1 mM, Figure 2B, left), but inactivated or weakly activated by choline (10 mM, Figure 2C, left). These observations suggest that α4β2-nAChRs are the predominant, functional nAChRs that mediate ID response in this part of putative SNc DAergic neurons. On the other hand, type IID receptors were strongly activated by ACh (1 mM, Figure 2A, right) and choline (10 mM, Figure 2C, right), but only weakly activated by RJR-2403 (0.1 mM, Figure 2B, right), indicating α7-nAChRs are the predominant, functional nAChRs that mediate IID response in this set of putative SNc DAergic neurons. As summarized in Figure 2D, only 14 of 61 (23%) recorded putative DAergic neurons mainly express type IID nAChRs (this part of neurons were named as type IID neuron), while 47 of 61 (77%) recorded putative DAergic neurons mainly express type ID nAChRs (this part of neurons were termed as type ID neuron). These results suggest that both α4β2- and α7-nAChRs are expressed on the cell body of the putative DAergic neurons freshly dissociated from rat SNc, and α4β2-nAChRs are the major nAChR subtype (Figure 2D).
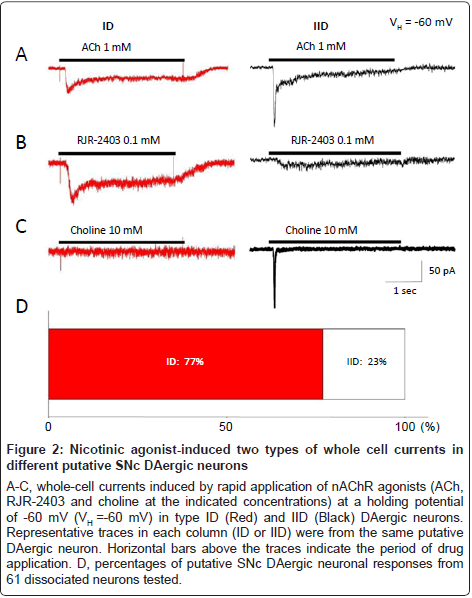
Figure 2:Nicotinic agonist-induced two types of whole cell currents in different putative SNc DAergic neurons
A-C, whole-cell currents induced by rapid application of nAChR agonists (ACh, RJR-2403 and choline at the indicated concentrations) at a holding potential of -60 mV (VH =-60 mV) in type ID (Red) and IID (Black) DAergic neurons. Representative traces in each column (ID or IID) were from the same putative DAergic neuron. Horizontal bars above the traces indicate the period of drug application. D, percentages of putative SNc DAergic neuronal responses from 61 dissociated neurons tested.
Modulation of GABAA receptor function by nicotine in SNc DAergic neurons
Then, we further examined the nicotinic modulation of GABAA receptor function by a smoke-relevant concentration of nicotine (500 nM). Under voltage-clamp conditions (VH=-60 mV), whole-cell currents were reliably obtained by fast application of exogenous GABA (10 μM, Figure 3A, Left) and were dramatically suppressed to 2.3 ± 1.4% of the control levels (Figure 3A, middle, Figure 3F, p<0.001, n=5 neurons from 4 rats) by 10 μM bicuculline, a competitive GABAA receptor antagonist suggesting that somatodendritic GABAA receptor mediated GABA-induced whole-cell currents. We further evaluated effects of 500 nM nicotine on GABAA receptor-mediated currents in identified type ID and IID SNc DAergic neurons by co-application of 500 nM nicotine plus 10 μM GABA. Compared to the controls (10 μM GABA alone, Figure 3E, left), nicotine did not show any significant effects on the peak amplitude of the whole-cell currents evoked by 10 μM GABA (Figures 3E middle and 3F, 100.5 ± 1.2%, p=0.72 vs. controls, 8 cells from 8 rats) in type IID neurons (expressing α7-nAChRs). However, in type ID DAergic neurons (expressing α4β2-nAChRs), 500 nM nicotine significantly and reversibly increased the amplitude of GABA-induced whole-cell currents to 172.5 ± 17.2% of the control levels (Figures 3B and 3F, p<0.001, n=19 neurons from 16 rats) in 63% neurons tested (19 of 30 recorded neurons, Figure 3G). We defined this type of neurons as subtype IDa. In 27% ID neurons tested (8 of 30), 500 nM nicotine dramatically and reversibly decreased the amplitude of 10 μM GABA-induced whole-cell currents to 55.5 ± 10.0% of the control levels (Figure 3C and 3F, p<0.05, n=8 neurons from 7 rats, Figure 3G). We defined these neurons as subtype IDb. In the remaining 10% (3 of 30 recorded neurons, Figure 3G) of the type ID neurons, 500 nM nicotine (Figure 3D) did not show significant effect on the amplitude of the whole-cell currents induced by 10 μM GABA (100.2 ± 2.0% of control, p=0.93, 3 neurons from 3 rats, Figure 3F). These neurons were defined as subtype IDc. Together, these findings demonstrate that 500 nM nicotine significantly regulates GABAA receptor-mediated currents in the majority of putative SNc DAergic neurons in complex manners.
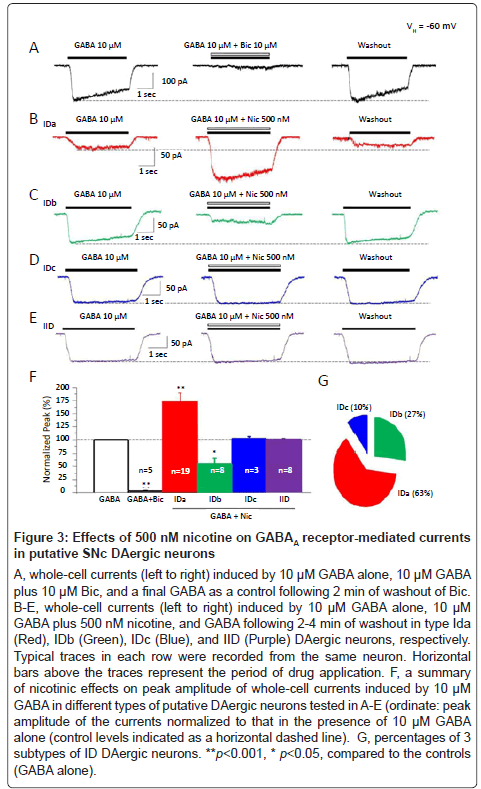
Figure 3:Effects of 500 nM nicotine on GABAA receptor-mediated currents in putative SNc DAergic neurons
A-C, whole-cell currents induced by rapid application of nAChR agonists (ACh, RJR-2403 and choline at the indicated concentrations) at a holding potential of -60 mV (VH =-60 mV) in type ID (Red) and IID (Black) DAergic neurons. Representative traces in each column (ID or IID) were from the same putative DAergic neuron. Horizontal bars above the traces indicate the period of drug application. D, percentages of putative SNc DAergic neuronal responses from 61 dissociated neurons tested.
To determine concentration-response relationships for nicotinic modulation of GABAA receptor function, a broad range of concentrations of nicotine (from 1 nM to 10 μM) plus 10 μM GABA were co-applied to type IDa (Figure 4A), IDb (Figure 4B), and type IID (Figure 4C) DAergic neurons, respectively. As show in Figure 4D, no significant changes modulation in magnitude were detected in all IDa (p>0.05, ANOVA, n=4), IDb (p>0.05, ANOVA, n=4), and IID neurons tested (p>0.05, ANOVA, n=5) although the concentrations of nicotine have been dramatically increased from 1 nM to 10 μM. These data indicate that nicotine exerts its modulatory effects on GABAA receptor function in a concentration-independent manner.
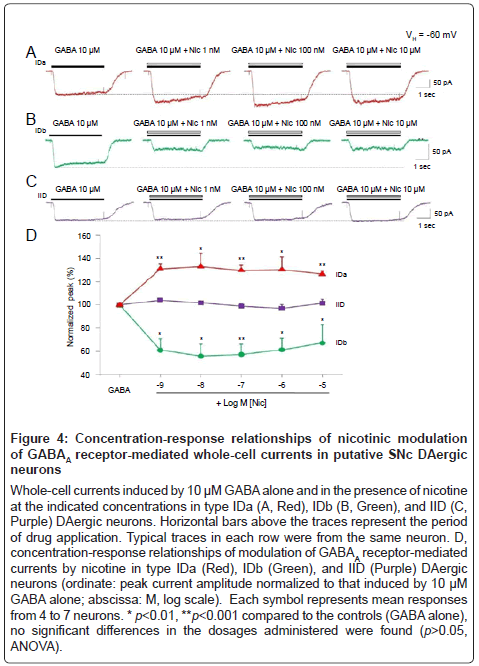
Figure 4:Concentration-response relationships of nicotinic modulation of GABAA receptor-mediated whole-cell currents in putative SNc DAergic neurons
A-C, whole-cell currents induced by rapid application of nAChR agonists (ACh, RJR-2403 and choline at the indicated concentrations) at a holding potential of -60 mV (VH =-60 mV) in type ID (Red) and IID (Black) DAergic neurons. Representative traces in each column (ID or IID) were from the same putative DAergic neuron. Horizontal bars above the traces indicate the period of drug application. D, percentages of putative SNc DAergic neuronal responses from 61 dissociated neurons tested.
The next question we asked was whether DHβE, a selective α4β2- nAChR antagonist, could prevent nicotinic modulation of GABAA receptor function since nicotinic modulation of GABAA receptor function only was observed in type ID neurons, which mainly express α4β2-nAChRs. The results showed that 500 nM nicotine significantly increased the amplitude (231 ± 29%, p<0.01 vs. control, n=4) of the whole-cell currents induced by 10 μM GABA (Figure 5Aii), and with 4 min pretreatment with 1 μM DHβE (Figure 5Aiii), this nicotinic enhancement of GABAA receptor-mediated current did not change significantly (peak amplitude of the currents was 230 ± 33% of control, p< 0.01, n=4, Figure 5B). Taking in consideration that α4β2-nAChRs could be desensitized rapidly when they were long exposure to 500 nM nicotine [48], the same recorded neurons were further incubated with 500 nM nicotine for 4 min, then co-application of 500 nM nicotine + 10 μM GABA still increased the amplitude of GABAA receptor-mediated currents to 214 ± 22% of the control levels (p< 0.01, n=4, Figure 5B). Figure 5B summarized the effects of 4 min pretreatment with either DHE or nicotine and showed that these manipulations did not affect nicotinic potentiation of GABAA receptor mediated whole-cell currents (p>0.05, ANOVA) in all neurons tested. These results suggest that nicotinic modulation of somatodendritic GABAA receptor function is likely not mediated through α4β2-nAChRs.
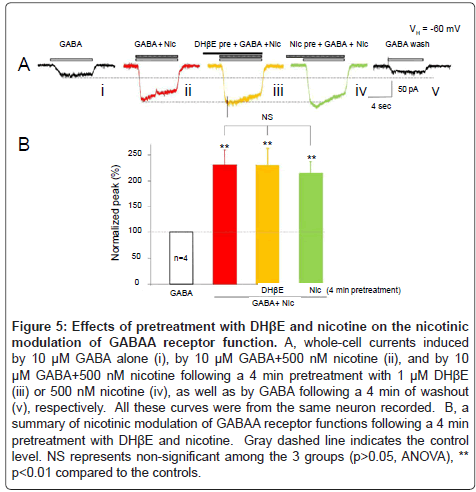
Figure 5: Effects of pretreatment with DHβE and nicotine on the nicotinic modulation of GABAA receptor function. A, whole-cell currents induced by 10 μM GABA alone (i), by 10 μM GABA+500 nM nicotine (ii), and by 10 μM GABA+500 nM nicotine following a 4 min pretreatment with 1 μM DHβE (iii) or 500 nM nicotine (iv), as well as by GABA following a 4 min of washout (v), respectively. All these curves were from the same neuron recorded. B, a summary of nicotinic modulation of GABAA receptor functions following a 4 min pretreatment with DHβE and nicotine. Gray dashed line indicates the control level. NS represents non-significant among the 3 groups (p>0.05, ANOVA), ** p<0.01 compared to the controls.
Nicotine did not significantly modulate glutamate- and glycine-induced whole-cell currents in SNc DAergic neurons
To test the possibility that smoking-relevant concentration of nicotine may also have effects on other ion channels, we also examined the effects of nicotine on whole-cell currents induced by either 100 μM glutamate (Glu, Figure 6A) or 30 μM glycine (Gly, Figure 6B) in putative SNC DAergic neurons, respectively. Results showed that 500 nM nicotine did not affect the whole-cell currents induced by Glu or Gly (Figure 6C, Glu: 99.9 ± 0.9%, p=0.9 vs. control, 5 cells from 5 rats; Gly: 99.7 ± 1.9%, p=0.9 vs. control, 8 cells from 7 rats). These results suggest that nicotine (500 nM) selectively produces its modulatory effects on GABAA receptor function, but not on glutamate or glycine receptor function, in SNc DAergic neurons.
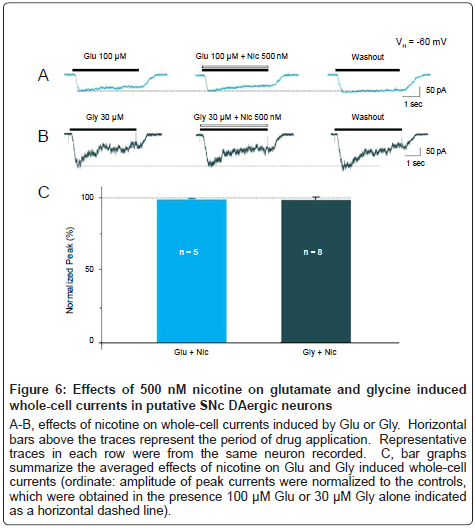
Figure 6: Effects of 500 nM nicotine on glutamate and glycine induced whole-cell currents in putative SNc DAergic neurons
A-B, effects of nicotine on whole-cell currents induced by Glu or Gly. Horizontal bars above the traces represent the period of drug application. Representative traces in each row were from the same neuron recorded. C, bar graphs summarize the averaged effects of nicotine on Glu and Gly induced whole-cell currents (ordinate: amplitude of peak currents were normalized to the controls, which were obtained in the presence 100 μM Glu or 30 μM Gly alone indicated as a horizontal dashed line).
In the present study, we provide direct evidence that smokingrelevant level of nicotine modulates somatodendritic GABAA receptor function in putative DA neurons of rat SNc. The mechanistic study further demonstrates that the nicotinic modulation of GABAA receptor function occurs in complex manners, which may depend on nAChR subtypes or GABAA receptor subtypes or other nonnAChR mechanisms. Finally, we find that GABAA receptor on SNc DA neuronal somato is likely a specific target for nicotinic modulation since the similar concentration of nicotine does not alter glutamate or glycine receptor function. Taken together, our data provide insights into understanding of how nicotinic modulation of SNc DA neuronal function, which may not only help to uncover the mechanisms of nicotinic protection in PD pathogenesis, but also open a new window to develop an effective therapeutic strategy for PD treatment.
Previous studies using single cell RT-PCR [49] and doublelabeling in situ hybridization [31] have demonstrated that at least eight nAChR subunits (α3-7 and β2-4) are well expressed in rodent SNc DAergic neurons. Consistent with these findings, we observed that functional nAChRs are present on all recorded DAergic neurons acutely dissociated from rat SNc. Furthermore, the present study has extended those previous observations by using whole-cell patch-clamp recordings and nAChR subunit selective agonists and demonstrated that two types of functional nAChRs, choline-sensitive and RJR-2403- sensitive nAChRs, are expressed on SNc DAergic neurons. Similar to our previous description of nAChRs expressed on DAergic neurons in rat VTA [30], we termed choline-sensitive and RJR-2403-sensitive nAChRs as IID (α7)- and ID (α4β2)-nAChRs in SNc DAergic neurons, respectively. Our electrophysiological data showed (Figure 2D) that functional α7-nAChRs are only expressed in 23% of tested putative DAergic neurons (type IID neuron), while functional α4β2-nAChRs are present in 77% of recorded putative DAergic neurons (type ID neuron) dissociated from the SNc. Thus, functional α4β2-nAChRs are the majority nAChRs richly expressed in SNc DAergic neurons. These observations are in well agreement with the previous report that β2 subunit containing-nAChRs are richly expressed in both the SNc and VTA, while α7-nAChRs have a lower expression level, in particular in SNc DAergic neurons [50].
It is well documented that nicotine or endogenous ACh either can directly enhance neuronal excitation via membrane depolarization mediated by postsynaptic nAChRs [30,50], or can produce its modulatory effects through nAChRs located on presynaptic terminals [28,37,51-53]. For example, nAChRs located on presynaptic DAergic nerve terminals may directly modulate dopamine D2/D3 receptor function [54,55], while somatodendritic nAChRs can regulate the function of GABAA receptors, which are expressed on the cell body of chick ciliary ganglion neurons and CA1 interneurons in hippocampal slices [35,36]. These findings suggest a potent functional crosstalk between nAChRs and other receptors including GABAA receptors.
Our present electrophysiological studies further expand the above observations to GABAA receptors in SNc DAergic neurons. We found that nicotine either enhances or inhibits whole-cell current-mediated by GABAA receptors in most of type ID DAergic neurons, which express functional α4β2-nAChRs, but nicotine has no significant effect on GABAA receptor-mediated whole-cell currents in all type IID DAergic neurons, which express functional α7-nAChRs. These findings suggest a potential functional crosstalk between α4β2-nAChRs and GABAA receptors at postsynaptic level in DAergic neurons freshly dissociated from rat SNc. Importantly, our studies further reveal that smokingrelevant concentrations of nicotine mainly increases the amplitude of GABAA receptor-mediated whole-cell currents in the majority (Figure 3G, 63%) of putative SNc DAergic neurons expressing α4β2-nAChRs (subtype IDa neurons), which indicates that nicotine effectively boosts the function of GABAA receptors expressed on DAergic neurons in the SNc. In the CNS, GABAA receptors are responsible for mediating inhibitory effects of endogenous GABAergic inhibitory system [56]. Activation of GABAA receptors tends to hyperpolarize the postsynaptic membrane due to the reversal potential for chloride ion (Cl-) in most neurons of adult animals and those older than P10-P14 is more negative than the resting membrane potential [57], which will lead to the postsynaptic neurons get less prone to excitatory neurotransmitters and generate action potential [56]. our observations provided the first evidence that low concentrations of nicotine obtained during smoking is sufficient to enhance postsynaptic GABAA currents onto DAergic neurons in the SNc, which may in turn reduce the excitability of SNc DAergic neurons because of these more hyperpolarization of GABAergic responses. In other words, nicotine may produce its neuroprotective effects via hyperpolarizing SNc DAergic neurons. We speculated that nicotine induced increases in GABAergic transmission may be a possible novel mechanism to explain how nicotine protects against the loss of SNc DAergic neurons [58-60], and this effect is most likely mediated through α4β2-nAChRs related mechanisms.
Importantly, we found that nicotine at very low concentrations (eg. 1 nM-100 nM), which are ineffective to activate postsynaptic nAChRs (data not shown), significantly enhance GABAA receptor function, while higher concentrations of nicotine do not produce even stronger modulatory effect (Figure 4). This concentration–independent modulation of GABAA receptor function by nicotine suggests that conventional receptor ligand model could not be used to interpret the phenomenon observed here. Furthermore, the nicotinic enhancement of GABAA receptor function could not be prevented by DHβE, though it was supposed that nicotine should produce its modulatory effects via functional α4β2-nAChRs expressed on the neuron. These results echo the findings by Zhang and colleagues that the effect of nicotine against β-amyloid-mediated neurotoxicity could not be blocked by nAChR antagonists [61], suggesting that nicotine may exert its modulatory effects on GABAA receptor-induced currents via a nAChR-independent pathway, or additional mechanisms may exist. For example, SNc DA neurons may express different subtypes of GABAA receptors [62-64] and the diversity of GABAA receptors may underlie the complex modulation by nicotine. This hypothesis needs to be tested in the further study. Further evidence supporting receptor-independent effects of nicotine comes from the finding showing that no significant alterations in the modulatory effects of nicotine on GABAA receptor-mediated currents were observed after 4 min pretreatment with 500 nM nicotine since it is well known that α4β2-nAChRs are easy to be desensitized following relative long exposure to low concentrations of nicotine [48,50]. Importantly, this novel action produced by nicotine may be particularly important for nicotine to produce its neuroprotection observed in smokers due to the following considerations: (a) nicotine only can significantly but shortly enhance GABA release from GABAergic terminals (boutons) through presynaptic α4β2-nAChRs [37] in the SNc because of the rapid desensitization resulted from chronic exposure to nicotine after tobacco smoking [48,50]. Thus, under chronic smoking conditions, this presynaptic modulation of GABA release could not last a long time. (b) The disinhibition produced by nicotine even can increase the excitability of DAergic neurons [41,62,63]. (c) We find that nicotine directly potentiates GABAA receptor-mediated postsynaptic whole-cell currents through postsynaptic rather than presynaptic mechanisms resulting in a hyperpolarization and reduction of excitability of SNc DAergic neurons. Importantly, pretreatment of nicotine does not affect nicotinic modulation of GABAA receptor function, which might be a potential mechanism underlying nicotinic neuroprotection.
In contrast to previous reports showing postsynaptic α7-nAChRs directly inhibit GABAA receptor function [35,36], we demonstrate that nicotine has no significant effect on GABAA receptor-mediated wholecell currents in those type IID DAergic neurons, which mainly express functional postsynaptic α7-nAChRs. Our results, meanwhile, also different from the previous report that nicotine does not significantly alter postsynaptic currents induced by exogenous GABA (30-50 μM) [64]. These contradictions could reflect real differences in preparations used in these experiments (eg. DAergic neurons from the SNc vs. neurons from the lateral spiriform nucleus, ciliary ganglion, and hippocampal interneurons) from different species (eg. Wistar rat vs. embryonic chick, or mice). Therefore, it remains to be determined whether the modulatory effects of nicotine on postsynaptic GABAA receptor function observed in this study are representative of the relationship in other regions of the brain or in tissue from other species.
Another question still needs to be answered is why nicotine could reduce GABAA receptor-mediated currents in subtype IDb neurons. One possibility is that the modulatory effects of nicotine also depend on the subunit composition of GABAA receptors since several GABAA receptor subunits (α2, α3, α4, β1, β3 and β2) have been found in mice [65] and human [66] SNc DA neurons. In addition, our technical limitation only lets us study the preparations from young animals (from P14 to P21). It is important, however, to further determine whether these kinds of nicotinic modulation could be preserved in adult or even aged animals. Thus, the significance of the nicotinic modulation of postsynaptic GABAA receptor function is potentially far-reaching and further studies are still needed.
The present study found that functional α4β2-nAChRs and α7- nAChRs are expressed on single DAergic neurons dissociated from rat SNc, which are termed as type ID and IID DAergic neurons, and the majority of the SNc DAergic neurons mainly express α4β2-nAChRs. Nicotine significantly enhances GABAA receptor-mediated currents in majority of type ID DAergic neurons (expressing α4β2-nAChRs), but not in those type IID DAergic neurons (expressing α7-nAChRs), indicating that nicotinic modulation of somatodendritic GABAA receptors occurs in α4β2-nAChRs -containing DAergic neurons in rat SNc. The concentration-independent nicotinic modulation could not be prevented by α4β2-nAChRs antagonist, suggesting that some novel nicotinic receptor-independent mechanisms may be involved in the nicotinic effect. Our findings that nicotinic enhancement of postsynaptic GABAA receptor function in SNc DAergic neurons, at least in part, may help to understand the neuroprotective effects produced by nicotine in PD patients through tobacco smoking although the detailed mechanisms are still need to be further investigated.
This work has been supported by IMHR seed fund, ADCRC research grant, Philip Morris External Research Program, the Barrow Neurological Foundation, and NIH DA15389. Authors thank Dr. Jionglong Tu for his assistance for TH staining.