Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 0, Issue 0
Ventral tegmental area dopamine (DA) and GABA neurons express nicotinic acetylcholine receptor (nAChR) subtypes, whose net activation results in enhancement of DA release in the nucleus accumbens (NAc). This effect decreases after repeated nicotine (NIC) treatment via desensitization. We evaluated the effects of acute NIC on glutamate decarboxylase (GAD67)-positive GABA neurons in the VTA of green fluorescent protein (GFP) knockin (GAD-GFP) mice, and determined the expression of selected nAChR subunits in VTA GABA neurons. In vivo, tachylphylaxis accrued to repeated systemic, but not local administration of NIC. Microelectrophoretic application of NIC and the α7 nAChR partial agonist JN403 markedly enhanced the firing rate of VTA GABA neurons. This activation was suppressed by intraperitoneal administration of the α7 nAChR antagonist methyllycaconitine (MLA, 1 mg/kg), or the glutamate (GLU) NMDA receptor antagonist APV (1 mg/kg), but not by the non-selective non-competitive antagonist mecamylamine (MEC, 1 mg/kg). In patch clamp studies in the slice preparation, the α7 nAChR agonist choline (1-10 mM), in the presence of the muscarinic cholinergic antagonist atropine (50 μM), enhanced sEPSC and mini-EPSC frequency, but not amplitude, which was blocked by MLA (0.5 μM). JN403 (0.1-1 μM) enhanced evoked EPSCs, without affecting membrane currents of VTA GABA neurons. In patch clamp studies in dissociated VTA GABA neurons, choline+atropine failed to induce whole-cell current responses in most cells tested. Single cell RT-qPCR revealed that most GABA neurons did not express α7 nAChRs. Together, this indicates that NIC excites VTA GABA neurons via α7 nAChRs located on GLUergic terminals.
<Keywords: VTA; GABA; Dopamine; Nicotine; Glutamate; Nicotinic receptor
The alkaloid nicotine (NIC) is the major contributor to the maintenance of tobacco use [1]. The rewarding effects of nicotine have been tied to the mesolimbic dopamine (DA) system, which primarily consists of projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) [2,3]. Activation and desensitization of the various nicotinic acetylcholine receptors (nAChRs) in the mesolimbic DA system may be crucial factors underlying the effects of NIC on the VTA [4,5] and NAc [6].
The majority of endogenous cholinergic inputs into the VTA innervate GABA neurons [7,8]. Both GABA and DA neurons can be activated by NIC [5,9]. GABA neurons express α4 and β2 subunits, which can be blocked by the non-selective, non-competitive antagonist mecamylamine, or the relatively more selective competitive antagonist dihydro-β-erythroidine (DHβE) [5]. The acute effects of NIC in the VTA, predominantly affect GABA neurons. The most important nAChR subtypes associated with these cells (i.e. α4β2-containing nAChRs) desensitize rapidly after the action of NIC, leading to an excitation of DA neurons, through removal of the inhibitory influence of GABA.
In the VTA, NIC also modulates glutamate (GLU) release. Dopamine and GABA neurons in the VTA receive GLUergic input from the prefrontal cortex (PFC), providing the major excitatory control of the VTA, and ultimately DA release in the NAc [10-15]. Nicotine receptors on GLUergic terminals in the VTA are mainly homomeric α7 nAChRs located presynaptically [4], and its stimulation enhances the release of GLU on DA and GABA neurons [16,17]. NIC is present in the brain of smokers, about 10-20 sec after absorption [18,19], reaching blood concentrations between 250 and 500 nM during cigarette smoking [20]. These chronic concentrations are sufficient to desensitize α4β2 nAChRs. Alpha-7 nAChRs, on the other hand, are less susceptible to the agonistic action of NIC and require higher doses to become desensitized, and recover more rapidly than other nAChRs [21]. In other words, during smoking, the NIC brain concentration is high enough to excite DA neurons.
There is a growing interest in studying the role of VTA GABA neurons in drug abuse, in particular, NIC addiction. For example, a recent paper has shown that the concerted activation of VTA DA and GABA systems is necessary for the reinforcing actions of NIC [22]. Nicotine increases GABA neuron firing rates directly, by acting on specific nAChR subtypes [4]. However, the identification of GABA neurons has previously been based on electrophysiological criteria. Thus, the aim of our study is to evaluate the effects of NIC, as well as the expression of nAChR subtypes on firing rate and GLU synaptic transmission, in a homogeneous subpopulation of GABA neurons in the VTA of glutamic acid decarboxylase green fluorescent protein (GAD-GFP) mice, so that they can be evaluated unambiguously. We hypothesized that VTA GABA neurons would be excited by NIC via α4β2 nAChRs on cell bodies and α7 nAChRs on presynaptic GLUergic terminals. In vivo and in vitro studies were performed to establish the pharmacological mechanisms by which NIC affects VTA GABA neurons, and the physiological and molecular relevance of this action.
Animal subjects
Male CD-1 (white albino) mice were housed four to a cage from the time of weaning (P25), with ad libitum access to food and water. Their room temperature was controlled (22-25°C), and maintained on a reverse 12 hr light/dark cycle with lights ON from 8 PM to 8 AM. Animal care, maintenance and experimental procedures were in accordance with the Brigham Young University Animal Research Committee, and met or exceeded National Institutes of Health guidelines for the care and use of laboratory animals. Female CD-1 wild-type mice were bred with male CD-1 glutamate decarboxylase-67 (GAD67)-green fluorescent protein (GFP67) knock-in mice (GAD-GFP mice; [23]). The offspring from these crossings were heterozygous GAD-GFP mice. GAD-GFP+ offspring were distinguished from GAD GFP- offspring by gross inspection between PND1-7, with strong illumination and GFP optics. The GAD GFP mice afforded us the ability to positively identify and record from GAD65-positive GABA neurons in the VTA of horizontal brain slices via fluorescence microscopy.
Single-unit recordings in anesthetized mice
Extracellular potentials in Isoflurane (1%) anesthetized adult 30-40 g male GAD-GFP mice were recorded by a single 3.0 M NaCl filled micropipette (1-3 MΩ; 1-2 μm inside diameter), or by a single recording micropipette cemented 10-20 μm distal to a 4-barrel micropipette (20- 60 MΩ resistance) for drug iontophoresis, and amplified and filtered with a MultiClamp 700A programmable amplifier (Axon Instruments, Union City, CA). Microelectrode assemblies were oriented into the VTA (from bregma: 5.6-6.5 posterior (P), 0.5-1.0 lateral (L), 7.0-8.5 ventral (V)), with a piezoelectric inchworm microdrive (Burleigh, Fishers, NY). Single-unit activity was filtered at 0.3-10 kHz (-3dB), and displayed on Tektronix 2200 digital oscilloscopes. In some experiments, square-wave constant current pulses (50-1000 μA; 0.15 msec duration; average frequency, 0.1Hz) were generated by an IsoFlex constant current isolation unit controlled by a MASTER-8 Pulse Generator (AMPI, Israel), or by computer. Extracellularly recorded action potentials (min 5:1 signal-to-noise ratio) were discriminated with a WPI-121 (Sarasota, Fl) spike analyzer, and converted to computer-level pulses.
Characterization of VTA GABA neurons in vivo
All neurons classified as VTA GABA neurons in vivo were located in the VTA, and met the criteria established in previous studies for spike waveform characteristics in rats [24-26], and in mice [27,28]. Presumed VTA GABA neurons were characterized by short-duration (<200 μsec; measured at half-peak amplitude of the spike), initially negative-going, relatively fast-firing (10-80 Hz), non-bursting spikes. In some experiments, they were further identified by short latency (i.e. 2-5 msec) spike activation via single stimulation, and multiple spiking, following high-frequency (10 pulses, 200 Hz) stimulation of the internal capsule [24-26,29].
Analysis of single-unit recordings in vivo
Single-unit potentials, discriminated spikes and stimulation events in vivo were captured by National Instrument’s NB-MIO-16 digital I/O and counter/timer data acquisition boards (Austin, TX), and processed by customized National Instruments LabVIEW software in Macintoshtype computers. Potentials were digitized at 20 kHz and 12-bit voltage resolution. For single-unit activity, all spikes were captured by computer and time stamped. Spontaneous firing rates were determined on- and off-line from ratemeter records by rectangular integration over a 5 min epoch, typically 5 min before, and at designated intervals after drug injection. Peri-stimulus and interval-spike histograms were generated off-line using IGOR Pro (WaveMetrics, Lake Oswego, OR).
Characterization of VTA GABA neurons in vitro
In GAD-GFP knock-in mice [23], GABA neurons were studied in horizontal brain slices with the aid of fluorescence microscopy. The VTA was visualized by first locating the substantia nigra reticulata (SNr) in the horizontal slice preparation, under low power (4X) fluorescence illumination. The SNr has a characteristic glow under low magnification with GFP fluorescence optics, likely due to dense GABA terminal innervation. Substantia nigra compacta (SNc) was then identified medial to SNr. GABA neurons in the VTA were studied by visualizing GAD+ neurons in an area medial to the glowing SNr, posterior to the fasciculus retroflexus and mammillothalamic tract, and anterior to the decussation of the superior cerebellar peduncle [27,28]. Neurons in the VTA of GAD-GFP mice that did not fluoresce but exhibited a non-cation specific inward rectifying current (Ih), in combination with relatively low input resistance and regular, slow spike activity were assumed to be DA neurons [25,27,28,30,31].
Drug preparation and administration
For iontophoretic experiments in vivo, NIC (nicotine hydrogen tartrate salt; Sigma-Aldrich, St Louis, MO) or JN403 was dissolved in 1 mL of distilled water for 4-barrel micropipette studies (0.7 mM), or 1 M KCl for single-barrel microelectrode studies (1-2 MΩ; 0.7 mM), and ejected with positive current (+40 nA), with an Axon Instruments MultiClamp 700A amplifier in current clamp mode. For systemic experiments, NIC or the NMDA antagonist (+/-)-2-amino-5- phosphonovaleric acid (APV; Ascent Scientific) were dissolved in 0.9% saline and administered intravenously through an indwelling jugular catheter. The NIC antagonists mecamylamine hydrochloride (MEC; Sigma-Aldrich), dihydro-beta-erythroidine (DHBE; Sigma-Aldrich), methyllycaconitine (MLA; Ascent Scientific) were dissolved in 0.9% saline and administered intraperitoneally.
Preparation of brain slices
GAD-GFP mice were anesthetized with Ketamine (60 mg/kg) and decapitated. The brains were quickly dissected and sectioned in ice-cold artificial cerebrospinal fluid (ACSF), bubbled with 95% O2/ 5% CO2. This cutting solution consisted of (in mM): 220 Sucrose, 3 KCl, 1.25 NaH2PO4, 25 NaH2CO3, 12 MgSO4, 10 Glucose, 0.2 CaCl2, and 0.4 Ketamine. VTA targeted horizontal slices (210 μm thick) were immediately placed into an incubation chamber containing normal ACSF at 34-35°C, bubbled with 95% O2/5% CO2 at 36°C consisting of (in mM): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 12 glucose, 1.5 MgSO4, 2 CaCl2, pH 7.3, and allowed to incubate for at least 45 minutes prior to being transferred to a recording chamber. Once transferred to a recording chamber with continuous normal ACSF flow (2.0 ml/min) maintained at 34-35°C throughout the experiment, the slices were then allowed to settle for an additional 15 to 30 minutes before recordings begins. These incubation and settling periods allowed cells to recover and stabilize. Cells were visualized for patching with either Nikon Eclipse FN1 or E600FN microscopes in the transmitted de Sénarmont Differential Interference Contrast (DIC)/infrared (IR) configuration (IR-DIC).
Whole-cell recordings in vitro
Electrodes pulled from borosilicate glass capillary tubes were filled with one of two types of pipette solutions. The pipette solution consisted of (in mM): 128 KCl, 20 NaCl, 0.3 CaCl2, 1.2 MgCl2, 10 HEPES, 1 EGTA, 2 Mg-ATP, 0.25 Na-GTP and 4.5 QX314 (pH 7.3) to block sodium channels intracellularly. Series resistance (Ra), typically 10 to 20 MΩ, and input resistance (Rm), typically 300 to 400 MΩ, were continuously monitored with a 10 mV 20 msec voltage step delivered at 0.1 Hz throughout each experiment, and only experiments that maintained stable Ra and Rm (less than 20% change) were included in this study. Synaptic events were filtered at 2 kHz using an Axon Instruments Multiclamp 700B amplifier, and digitized at 10 kHz using an Axon 1440A digitizer, and collected and analyzed using pClamp10 and Igor Pro (Wavemetrics: Oswego, OR) software packages. VTA GABA neurons were identified as described above and patched first with a gigaohm seal, and then the pipette was aspirated to break the membrane and achieve whole-cell recordings. To assure that spontaneous currents associated with relatively rapid VTA GABA neuron spiking would not confound the analysis of synaptic events, we performed a special protocol that would assure block of sodium currents. Within 1-2 min after going whole-cell, a voltage command waveform was administered, in order to induce high-frequency sodium currents (>200 Hz for 500 msec) in VTA GABA neurons, which facilitated the block of sodium channels with the use-dependent blocker QX-314 from inside the cell [32]. This protocol was repeated until sodium currents could no longer be elicited (typically only 2 trials blocked all sodium currents). Evoked and spontaneous EPSCs (sEPSCs) were recorded in the presence of 100 μM picrotoxin to block inhibitory synaptic transmission. Miniature EPSCs (mEPSCs) were isolated from spike-related events by addition of 0.5 μM TTX, or 500 μM lidocaine. To evoke EPSCs (eEPSCs), cells were stimulated at 0.1 Hz with a stainless steel-platinum/iridium concentric bipolar stimulating electrode placed ~ 100-300 μm rostral to the recording electrode. Evoked EPSCs were inward at the holding potential of -70 mV, and were completely blocked by 100 μM D-L 2-amino-5- phosphonopentanoic acid (APV) and 30 μM 6-cyano-23-dihydroxy-7- nitro-quinoxaline (CNQX). Evoked EPSCs amplitudes were calculated by taking the difference between the 1.0 msec window around the peak, and the 5.0 msec baseline window immediately preceding the stimulation artifact. Spontaneous EPSC activity amplitude and frequency was calculated the same for both sEPSCs and mEPSCs; the average amplitude or frequency during a 2 min period, 1 min following drug was normalized to the average amplitude or frequency from a 2 min window prior to drug.
Acutely dissociated neurons from the mouse VTA
Single neurons were acutely dissociated from the VTA of 2-3-weekold mice (from Charles River), following procedures as previously described [33,34]. Briefly, mice were anesthetized with isoflurane, and brain tissue was rapidly removed and immersed in cold (2-4°C) artificial cerebrospinal fluid (ACSF), which contained (in mM): 124 NaCl, 5 KCl, 24 NaHCO3, 1.3 MgSO4, 1.2 KH2PO4, 2.4 CaCl2, and 10 glucose; pH 7.4. From each mouse, two 400 μm coronal slices containing the VTA were cut using a vibratome (Vibratome 1000 Plus, St. Louis, MO). After cutting, the slices were continuously bubbled with 95% O2-5% CO2 at room temperature (22 ± 1°C), for at least 1 h in ACSF. Thereafter, the slices were treated with papain (6 mg/ml, Sigma-Aldrich) at 33°C for 60 min in ACSF. After enzyme treatment, the slices were washed and transferred back to well-oxygenated ACSF, for at least 15 min. The VTA was first identified using a stereomicroscope, and then micro-punched out from the slices using a well-polished needle (0.5 mm diameter). Each punched piece was then separately transferred to a 35-mm culture dish filled with well-oxygenated standard extracellular solution, which contained (in mM): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES; pH 7.4 (with Tris-base). Each punched piece was then dissociated mechanically using a fire-polished micro-Pasteur pipette, under an inverted microscope (Olympus IX-70, Lake Success, NY). The separated cells adhered to the bottom of the culture dish within 30 min. We used only VTA neurons that maintained their original morphological features of small- or medium-sized somata, with 2-4 thin, primary dendritic processes.
Perforated patch-clamp whole-cell recordings
Perforated-patch whole-cell recording techniques were employed [33,34]. Compared to performing conventional whole-cell recordings, this approach was crucial for obtaining stable nAChR responses from dissociated VTA neurons, presumably due to minimal perturbation of the intracellular environment. Pipettes (3-5 MΩ) used for perforatedpatch recordings were filled with intracellular recording solution, which contained (in mM): 140 K-gluconate, 10 KCl, 5 MgCl2, and 10 HEPES; pH 7.2 (with Tris-OH), freshly supplemented before use with amphotericin B to 200 μg/ml from a 40 mg/ml DMSO stock. The liquidjunction potential was 14 mV and calculated using Clamplex 9.2 (Axon Instruments, Foster City, CA), and corrections were made for junction potentials post-hoc. After tight seal (>2 GΩ) formation, about 5-20 min was required for conversion to the perforated-patch mode, and an access resistance of <60 MΩ was accepted to start the experiments. Series resistance was not compensated in this study. Data were acquired at 10 kHz, filtered at 2 kHz, digitized on-line (Digidata 1322 series A/D board, Axon Instruments), and displayed and stored on a PC computer. Rapid application of drugs was performed using a computer-controlled “U-tube” system that allowed for complete exchange of solution surrounding the recorded cell within 30 msec. Periods of drug exposure are specified in the text and figures or legends. When ACh or choline was used as a nAChR agonist, 1 μM atropine was routinely added to exclude actions mediated via endogenous muscarinic receptors, and this manipulation is known to have no clear effect on ACh-induced currents [33]. All experiments were performed at room temperature. The GABAergic neurons were initially identified based on neuronal electrophysiological properties. Compared to DA neurons, GABAergic neurons have fast spontaneous action potential firing (>6 Hz), shorter action potential duration (the duration at 50% spike amplitude <2 ms), no hyperpolarizing-induced current and insensitivity to DA. To further confirm these electrophysiologically identified GABAergic neurons, we delivered fluorescent dye (lucifer yellow; Sigma-Aldrich, St. Louis, MO; 1.0 mg/ml in the recording electrode) in some experiments. After conversion from perforated to conventional whole-cell recording mode, the dye was injected into the cytoplasm by a pulse (200 ms, 0.5 Hz) of hyperpolarizing current (1.0 nA), for 3 min. Labeled cells were visualized using epifluorescence microscopy and tyrosine hydroxylase (TH) staining post-hoc. In five cells tested, all electrophysiologically identified GABAergic neurons showed negative reaction for tyrosine hydroxylase staining.
Single-cell quantitative RT-PCR
Following electrophysiological characterization, GAD-GFP VTA GABA neurons in mature mice were aspirated with gentle suction under visual observation, and were immediately added to a reverse transcription (RT) reaction mixture. The iScript cDNA synthesis kit (BioRad) was used for a total volume of 10 μl per reaction. Reactions were run at 25.0°C for 8 minutes, 42.0°C for 60 minutes, and 70°C for 15 minutes in a C1000 thermal cycler (BioRad). A preamplification round of multiplex PCR was performed by adding diluted primers and iQ Supermix (BioRad) to the completed RT reaction, for a final volume of 30 μL. The reactions were cycled with a 95°C hot start step for 3 minutes, followed by 15 cycles of 95°C for 15 seconds, 57°C for 20 seconds, and 72°C for 25 seconds. Samples (1.5 μl) of the initial multiplex PCR was then used as substrate for each reaction in the subsequent real-time quantitative PCR. Real-time quantitative PCR using gene specific primers (TH forward primer: GGACAAGCTCAGGAACTATGC, TH reverse primer: GGTGTACGGGTCAAACTTCAC; 18s forward primer: GTGCATGGCCGTTCTTAGTTG, reverse 18s primer: GCCACTTGTCCCTCTAAGAAGTTG; α4 forward primer: TGGTCCTTGTCCGCTTTG, α4 reverse primer: CGTCATCATCTGGTTTTTCTCATC; α6 forward primer: TCACGGTGCATTTTGAATTG, α6 reverse primer: GGTCTCCATAATCTGGTTGACTTC; α7 forward primer: TTGCCAGTATCTCCCTCCAG, α7 reverse primer: CTTCTCATTCCTTTTGCCAG; β2 forward primer: CCTGAGGATTTCGACAATATGAAG, β2 reverse primer: GCATTGTTGTATAGAACCACATCTG; Connexin 36 forward primer: TGCAGCAGCACTCCACTATG, Connexin 36 reverse primer: ATGGTCTGCTCATCATCGTACAC; Invitrogen), with FAM-TAMRA TaqMan® probes (TH probe: CCCCATGTGGAATACACAGCGGAAGAG; 18s probe: TGGAGCGATTTGTCTGGTTAATTCCGATAAC; α4 probe: CTTGTCGATTGCTCAGCTCATTGATGT; α6 probe: CAATCACGCAACTGGCCAATGTG; α7 probe: ATGTACGCTGGTTCCCTTTTGATGTGC; β2 probe: TCCGACTCCCTTCTAAGCACATCTGGC; Connexin 36 probe: ATCCTGTTGACTGTGGTGGTGATCTTCC; Applied Biosystems) were performed using iQ Supermix (BioRad), with a CFX96 qPCR System (Bio-Rad). Samples were amplified in triplicate, together with a negative control for each subunit, which was an ACSF-only aspiration taken directly above the slice in an electrode that was previously placed in the slice. The amplification protocol was 95°C hot start for 3 minutes, followed by 50 cycles of 95°C for 15 seconds, 57°C for 20 seconds, and 72°C for 25 seconds. Cycle threshold (Ct) values were calculated using CFX Manager Software (BioRad). Amplification products from each sample were verified by gel electrophoresis using 4% agarose gels.
Statistical analyses
The results for control and drug treatment groups were derived from calculations performed on VTA GABA neuron spontaneous firing rate. A two-tailed student’s t test was invoked for comparison between control and treatment within subject. Comparison among individual means was made by Newman-Keuls post-hoc tests, following ANOVA. Further analyses of saline vs. nicotine dose-response data, or other repeated measures were made by Duncan’s new multiple-range test, to determine the source of detected significance in the ANOVAs. The criterion of significance was set at p<0.05.
Effects of systemic administration of nicotine on VTA GABA neuron firing rate in vivo
In anesthetized (Isoflurane) mice, the average firing rate VTA GABA neurons recorded in GAD-GFP mice in vivo was 31.5 ± 2.5 Hz (n=79), consistent with what we have reported previously in other studies in mice [28,35]. Intravenous administration of freebase NIC (0.2 mg/kg) markedly enhanced the firing rate of VTA GABA neurons recorded in GAD-GFP mice in vivo (Figure 1A). The rate of firing increased in about 20 sec and returned to baseline, approximately 3-4 min after the first NIC injection. However, repeated administration of systemic 0.2 mg/kg NIC at 5 min intervals resulted in a marked tachyphylaxis of the activating effects of NIC on VTA GABA neuron firing rate. Indeed, the third and fourth injections of NIC resulted in a significant decrease in the activation of firing rate compared to the first injection (Figure 1B; 1st vs 3rd: P=0.01, t(2,5) =3.3; n=6; 1st vs 4th: P=0.005, t(2,5)=4.8; n=6).
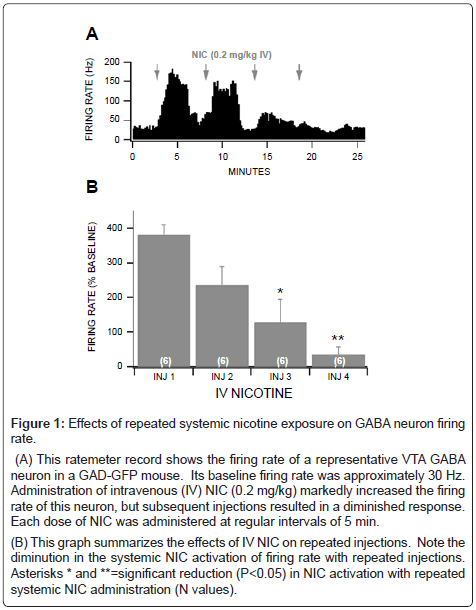
Figure 1: Effects of repeated systemic nicotine exposure on GABA neuron firing rate.
(A) This ratemeter record shows the firing rate of a representative VTA GABA neuron in a GAD-GFP mouse. Its baseline firing rate was approximately 30 Hz. Administration of intravenous (IV) NIC (0.2 mg/kg) markedly increased the firing rate of this neuron, but subsequent injections resulted in a diminished response. Each dose of NIC was administered at regular intervals of 5 min.
(B) This graph summarizes the effects of IV NIC on repeated injections. Note the diminution in the systemic NIC activation of firing rate with repeated injections. Asterisks * and **=significant reduction (P<0.05) in NIC activation with repeated systemic NIC administration (N values).
Pharmacology of local nicotine activation of VTA GABA neuron firing rate in vivo
In situ microelectrophoretic application of NIC (+40 nA) markedly enhanced the firing rate of VTA GABA neurons recorded in anesthetized GAD-GFP mice in vivo (330.7 ± 26.8%; P=5.6E-07, t(2,54)=5.9; n=55; Figures 2A and 2B). Although a robust tachyphylaxis was observed with repeated systemic NIC injections (Figure 1), it was surprising that there was no diminution of the NIC response with repeated, periodic (1.0 min ON, 1.0 min OFF) iontophoretic NIC current applications (Figure 2). The ability to repeatedly activate VTA GABA neurons, reliably with periodic NIC applications, justified the utility of antagonists to determine which nAChR subtype might be involved in NIC activation of VTA GABA neuron firing rate. Accordingly, as α7 nAChRs are typically located on GLU terminals [4], we evaluated the effects of intraperitoneal administration of the nonselective, non-competitive antagonist mecamylamine (MEC), the α7 nAChR antagonist methyllycaconitine (MLA), and the NMDA GLU receptor antagonist aminophosphonovalerate (APV), at the same dose level (1 mg/kg) on NIC activation of VTA GABA neuron firing rate in vivo. While MEC had no significant effects (P>0.05), MLA and APV significantly reduced NIC activation of VTA GABA neuron firing rate (MLA: P=0.001, t(2,9)=4.1; n=10; APV: P=0.04, t(2,6)=2.3; n=7; Figures 2A and 2B), compared to an isovolumic injection of saline. Since MLA suppressed NIC activation of VTA GABA neuron firing rate, we evaluated the effects of local application of the α7 nAChR partial agonist JN403. Microelectrophoretic application of JN403 (+40 nA) significantly (444.0 ± 64.1%; P=5.8E-05, t(2,23)=4.8; n=24) enhanced the firing rate of VTA GABA neurons. Intraperitoneal administration of MLA significantly reduced JN403-induced increase in VTA GABA neuron firing rate (P=0.008, t(2,4)=4.6; n=5; Figures 2C and 2D).
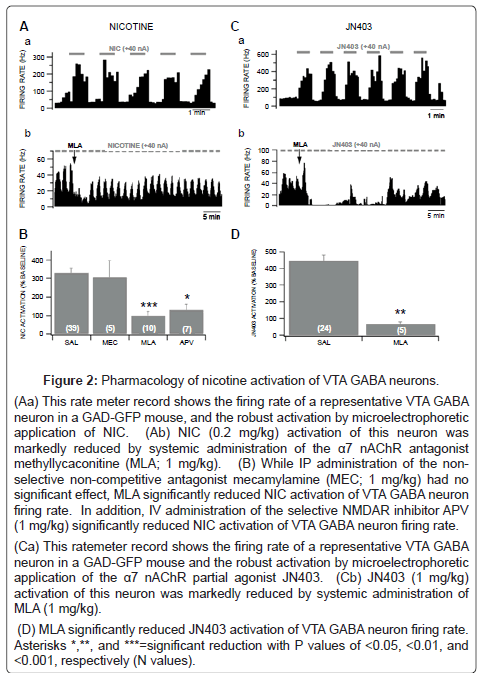
Figure 2: Pharmacology of nicotine activation of VTA GABA neurons.
(Aa) This rate meter record shows the firing rate of a representative VTA GABA neuron in a GAD-GFP mouse, and the robust activation by microelectrophoretic application of NIC. (Ab) NIC (0.2 mg/kg) activation of this neuron was markedly reduced by systemic administration of the α7 nAChR antagonist methyllycaconitine (MLA; 1 mg/kg).
(B) While IP administration of the nonselective non-competitive antagonist mecamylamine (MEC; 1 mg/kg) had no significant effect, MLA significantly reduced NIC activation of VTA GABA neuron firing rate. In addition, IV administration of the selective NMDAR inhibitor APV (1 mg/kg) significantly reduced NIC activation of VTA GABA neuron firing rate.
(Ca) This ratemeter record shows the firing rate of a representative VTA GABA neuron in a GAD-GFP mouse and the robust activation by microelectrophoretic application of the α7 nAChR partial agonist JN403. (Cb) JN403 (1 mg/kg) activation of this neuron was markedly reduced by systemic administration of MLA (1 mg/kg).
(D) MLA significantly reduced JN403 activation of VTA GABA neuron firing rate. Asterisks *,**, and ***=significant reduction with P values of <0.05, <0.01, and <0.001, respectively (N values).
Effects of α7 nicotinic receptor agonist on glutamatergic synaptic transmission to VTA GABA neurons
Based on in vivo studies (Figures 1 and 2), NIC markedly excites VTA GABA neurons, irrespective of the route of administration (i.e. systemic vs local). As the α7 nAChR antagonist MLA or the GLU antagonist APV suppressed NIC activation of VTA GABA neuron firing rate, we pursued ex vivo electrophysiology and pharmacology studies in the slice preparation, to determine the mechanism of action of NIC effects on excitatory synaptic transmission to VTA GABA neurons. We hypothesized that NIC was acting on α7 nAChRs on GLU terminals, enhancing the release of GLU to VTA GABA neurons. We tested the effects of the α7 nAChR agonist choline (10 mM) on VTA GABA neuron sEPSCs in the horizontal VTA slices. The average sEPSC frequency in VTA GABA neurons was 9.7 ± 1.7 Hz (n=20), and the average amplitude was 20.3 ± 1.53 pA (n=20). Superfusion of the slice with choline induced a slow inward current in VTA GABA neurons, and markedly enhanced sEPSC frequency (Figure 3Aa). However, as choline is also a muscarinic cholinergic agonist at this concentration level, we tested it in the presence of the muscarinic antagonist atropine (50 μM). The ability of choline-induced inward current was blocked by atropine (Figure 3Ab), but its ability to enhance sEPSCs remained unchanged (Figures 3Ac,d), albeit the enhancement was more transient than without atropine. Figure 3B summarizes the effects of choline on sEPSC frequency and amplitude. Choline alone significantly increased sEPSC frequency (P=0.001, t(2,19)=3.8; n=20) and amplitude (P=0.01, t(2,19)=2.8; n=20). Choline+atropine also significantly increased sEPSC frequency (P=0.01, t(2,14)=2.9; n=15), but not amplitude (P=0.16, t(2,14)=1.5; n=15). The effect elicited by choline+atropine was blocked by the α7 nAChR antagonist MLA (n=8; Figure 3B). Thus, action potential-dependent spontaneous GLU input to VTA GABA neurons in the slice preparation was modulated by activation of α7 nAChRs on GLU terminals, even when the muscarinic postsynaptic effects of choline were controlled. To further support our hypothesis that NIC enhances VTA GABA neuron firing rate via α7 nAChR-induced GLU release on GLUergic terminals, we tested the effects of choline+atropine on spontaneous miniature EPSCs (mEPSCs). The frequency of mEPSCs is a reliable measure of spontaneous GLU release, which we presumed would be sensitive to nAChR agonist modulation. The mEPSCs were recorded in the presence of tetrodotoxin (TTX; 500 μM, which blocked action potential-dependent release of GLU, and picrotoxin (PTX; 100 μM), which blocked inhibitory synaptic transmission. The averaged mEPSC frequency in VTA GABA neurons was 5.5 ± 1.5 Hz (n=10), and the averaged amplitude was 18.1 ± 1.6 pA (n=10). Superfusion of the slice with choline+atropine enhanced mEPSC frequency (Figure 3Ca,b). Figure 3D summarizes the effects of choline on mEPSC frequency and amplitude. Choline+atropine significantly increased mEPSC frequency (P=0.03, t(2,8)=2.6; n=9) and amplitude (P=0.04, t(2,8)=1.5; n=9), which was blocked by 0.5 μM MLA (n=8). For the same reasons listed above, although nAChR-mediated GLU input to VTA GABA neurons from the PFC may not be spontaneously operational in the horizontal slice, it could still be evoked by electrical stimulation. Thus, we tested the effects of the selective α7 nAChR partial agonist JN403 on EPSCs evoked by local stimulation (i.e. eEPSCs), which would effectively activate GLU terminals on VTA GABA neurons, irrespective of spontaneous action potential input. The average amplitude of eEPSCs in VTA GABA neurons was 56.1 ± 6.9 pA (n=30), and the average paired-pulse response (50 msec interstimulus-interval) was 99.4 ± 5.65% (n=30). Figure 4 shows the effects of NIC and JN403 on eEPSCs. While 1 μM NIC did not significantly alter eEPSC amplitudes (P=0.61, t(2,9)=0.53; n=10), superfusion of 1 μM JN403 significantly enhanced eEPSCs in VTA GABA neurons (P=0.02, t(2,11)=2.9; n=12).
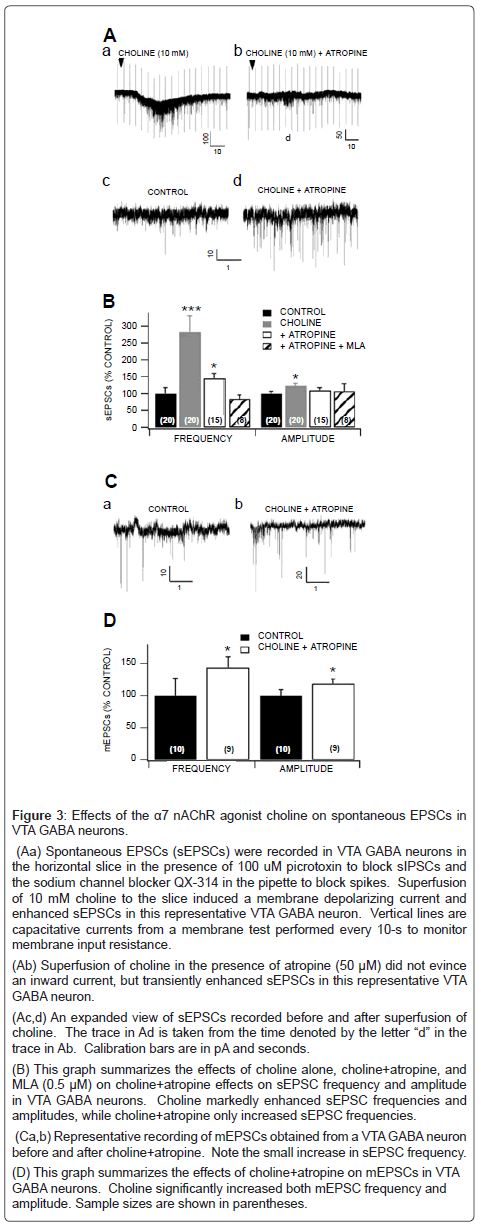
Figure 3: Effects of the α7 nAChR agonist choline on spontaneous EPSCs in VTA GABA neurons.
(Aa) Spontaneous EPSCs (sEPSCs) were recorded in VTA GABA neurons in the horizontal slice in the presence of 100 uM picrotoxin to block sIPSCs and the sodium channel blocker QX-314 in the pipette to block spikes. Superfusion of 10 mM choline to the slice induced a membrane depolarizing current and enhanced sEPSCs in this representative VTA GABA neuron. Vertical lines are capacitative currents from a membrane test performed every 10-s to monitor membrane input resistance.
(Ab) Superfusion of choline in the presence of atropine (50 μM) did not evince an inward current, but transiently enhanced sEPSCs in this representative VTA GABA neuron. (Ac,d) An expanded view of sEPSCs recorded before and after superfusion of choline. The trace in Ad is taken from the time denoted by the letter “d” in the trace in Ab. Calibration bars are in pA and seconds.
(B) This graph summarizes the effects of choline alone, choline+atropine, and MLA (0.5 μM) on choline+atropine effects on sEPSC frequency and amplitude in VTA GABA neurons. Choline markedly enhanced sEPSC frequencies and amplitudes, while choline+atropine only increased sEPSC frequencies.
(Ca,b) Representative recording of mEPSCs obtained from a VTA GABA neuron before and after choline+atropine. Note the small increase in sEPSC frequency.
(D) This graph summarizes the effects of choline+atropine on mEPSCs in VTA GABA neurons. Choline significantly increased both mEPSC frequency and amplitude. Sample sizes are shown in parentheses.
Lack of α7 nicotinic receptor function in dissociated VTA GABA neurons
In order to provide further evidence that NIC was acting on α7 nAChRs on GLU terminals, rather than on the somatodendritic area of VTA GABA neurons, we tested the effects of the α7 nAChR agonist choline on acutely-dissociated VTA GABA neurons. Under these conditions, dissociated single neurons have no presynaptic terminals/ boutons [34]. Thus, this single neuron model has only postsynaptic receptors. Under perforated whole-cell recording conditions in the voltage-clamp mode, bath-application of the α7 nAChR selective agonist choline (10 mM), via a U-tube, induced a small inward current at a holding potential of -60 mV (Figure 5A) in only a few cells tested. In the 32 cells tested, from 20 mice, only 6 neurons (18.7%, Figure 4B) exhibited choline responses with small amplitude of 10.2 ± 1.1 pA. Interestingly, ACh, GLU (1 mM), and GABA (0.1 mM) induced much larger inward current (Figure 5A-C). When choline induced inward currents in this voltage-clamp model, it showed little effect on AP firing in currentclamp recording mode, unlike ACh which markedly increased AP firing (Figure 5C). These results suggest that α7 nAChRs are not predominantly expressed on the somatodendritic area of GABA neurons, and the activation of α7 nAChRs on postsynaptic GABAergic neurons has little effect on GABAergic neuron firing.
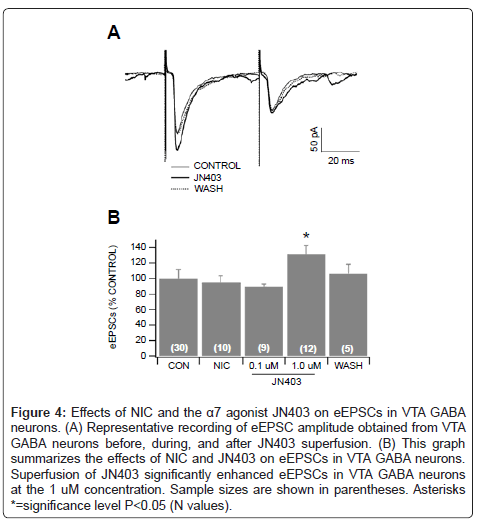
Figure 4: Effects of NIC and the α7 agonist JN403 on eEPSCs in VTA GABA neurons. (A) Representative recording of eEPSC amplitude obtained from VTA GABA neurons before, during, and after JN403 superfusion. (B) This graph summarizes the effects of NIC and JN403 on eEPSCs in VTA GABA neurons. Superfusion of JN403 significantly enhanced eEPSCs in VTA GABA neurons at the 1 uM concentration. Sample sizes are shown in parentheses. Asterisks *=significance level P<0.05 (N values).
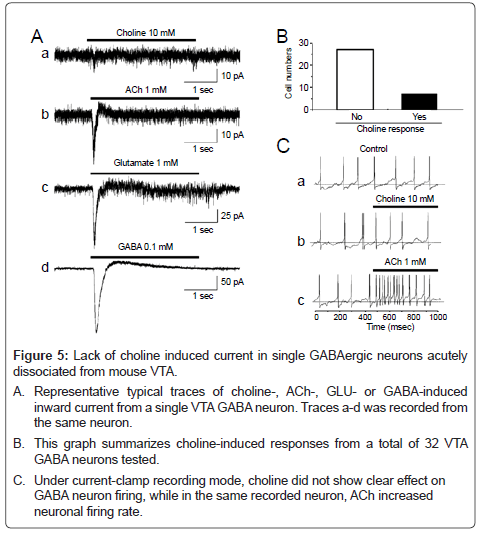
Figure 5: Lack of choline induced current in single GABAergic neurons acutely dissociated from mouse VTA.
A. Representative typical traces of choline-, ACh-, GLU- or GABA-induced inward current from a single VTA GABA neuron. Traces a-d was recorded from the same neuron.
B. This graph summarizes choline-induced responses from a total of 32 VTA GABA neurons tested.
C. Under current-clamp recording mode, choline did not show clear effect on GABA neuron firing, while in the same recorded neuron, ACh increased neuronal firing rate.
Lack of α7 nicotinic receptor expression in VTA GABA neurons
Performing quantitative real-time PCR on single VTA GABA neurons in GAD-GFP mice, we evaluated the expression of select nAChR mRNA transcripts. These cells were positively identified as GABA neurons using fluorescence microscopy, due to their expression of GFP. While many VTA GABA neurons tested expressed α4 and β2 nAChR subunits, as well as Cx36, a marker of VTA GABA neurons in GAD-GFP mice [25], few neurons expressed α6 subunits, and only two of the twenty-four neurons expressed α7 subunits (Figure 6).
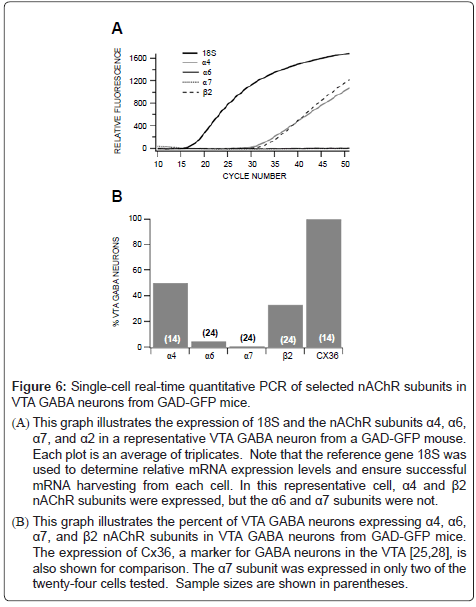
Figure 6: Single-cell real-time quantitative PCR of selected nAChR subunits in VTA GABA neurons from GAD-GFP mice.
(A) This graph illustrates the expression of 18S and the nAChR subunits α4, α6, α7, and α2 in a representative VTA GABA neuron from a GAD-GFP mouse. Each plot is an average of triplicates. Note that the reference gene 18S was used to determine relative mRNA expression levels and ensure successful mRNA harvesting from each cell. In this representative cell, α4 and β2 nAChR subunits were expressed, but the α6 and α7 subunits were not.
(B) This graph illustrates the percent of VTA GABA neurons expressing α4, α6, α7, and β2 nAChR subunits in VTA GABA neurons from GAD-GFP mice. The expression of Cx36, a marker for GABA neurons in the VTA [25,28], is also shown for comparison. The α7 subunit was expressed in only two of the twenty-four cells tested. Sample sizes are shown in parentheses.
We evaluated the activity of NIC on VTA GABA neuron firing rate in vivo to establish the pharmacological mechanisms involved in the acute effect elicited by NIC. Since systemic administration causes NIC circulation throughout the entire brain, NIC could be influencing neurons directly or indirectly through activation of nAChRs on afferents to the VTA, including GABA afferents to VTA GABA neurons from the NAc and ventral pallidum, as well as GLU inputs to VTA GABA neurons from the PFC that are sensitive to NIC [36,37]. In addition, there are putative ACh inputs from the habenula and pedunculopontine tegmental nucleus [38,39]. Thus, NIC systemic effects need to be evaluated in light of its possible net effects on GABA neurons and its afferents. However, since repeated administration of local NIC did not result in tachyphylaxis, one possible explanation is that a remote input to the VTA that is sensitive to NIC, in particular the PFC [40], might be responsible for the acute tolerance to NIC activation by systemic NIC. Admittedly, NIC activation of DA neurons in the VTA is known to induce DA release locally, which could have some effect on nearby neurons [40].
Nicotine activation, achieved with periodic in situ microelectrophoretic application, was found consistently and with profound magnitude, but did not diminish during current application or with repeated NIC exposures. Approximately 75% of VTA GABA neurons studied was activated by NIC in this manner. The ability of MLA to block NIC activation of VTA GABA neurons lends credence to the notion that α7 nAChRs play a crucial role in the activation of these neurons. The non-selective nAChR antagonist MEC had no effect on NIC activation of VTA GABA neuron firing rate, further supporting this proposition. Most importantly, local application of the potent α7 nAChR partial agonist JN403 [41], had consistent and robust excitatory effects on these neurons in vivo, as well. Indeed, JN403 excited VTA GABA neurons in all neurons tested without diminution, in response with repeated administrations. Its excitatory effect was also greatly diminished with systemic application of MLA, providing further evidence that NIC excites VTA GABA neurons via α7 nAChRs.
Glutamate projections from the PFC innervate both DA and GABA neurons [15,42], and often contain α7 nAChRs on GLUergic terminals [43]. Interestingly, α7 nAChRs were not associated with cholinergic synapses, consistent with their activation by a paracrine mode of ACh or choline delivery [43]. In order to evaluate the role of α7 nAChRs in local NIC activation of VTA GABA neurons, we tested the effects of the NMDA receptor antagonist APV. Nicotine activation was markedly reduced by systemic APV, suggesting that this increase in VTA GABA neuron firing rate was mediated by α7 nAChRs located on GLUergic terminals. Behavioral paradigms also support this electrophysiological evidence. MLA and APV have been shown to halt NIC’s rewarding effects, when microinfused with NIC into the VTA, during a conditioned place preference procedure in rats. As in our in vivo recordings in anesthetized mice, both α7 nAChR and NMDAR antagonists elicit similar outcomes.
Mansvelder et al. [5] has reported increases in GABA neuron firing rate with NIC in vitro. There is some concern, however, that the neurons they studied were not GABA neurons, as they relied on electrophysiological criteria alone (i.e. Ih current—DA neurons being Ih+ and GABA neurons being Ih−), which have been shown more recently to not be universally valid [30]. Accordingly, we have also observed repeatedly that GAD+ neurons in the VTA from GAD-GFP mice often evidence an appreciable Ih. Our in vitro data (Figure 3) takes advantage of visually identified GABA neurons in GAD-GFP mice in slice recordings and acutely dissociated neurons, to further support our understanding of NIC’s mechanism of action in the VTA. Our in vivo findings suggested that NIC actions on α7 nAChRs on GLUergic innervations in the VTA enhanced the activity of VTA GABA neurons. Only a small percentage of dissociated VTA GABA neurons were sensitive to choline and none of the VTA GABA cells in GAD-GFP tested positive for α7 nAChR transcripts, supporting a presynaptic locus for α7 effects on GLU terminals to VTA GABA neurons. However, there is evidence that some VTA DA and GABA neurons express α7 nAChRs [44]. We have previously shown that the firing rate of VTA GABA neurons is strongly mediated by corticotegmental NMDAR-mediated GLU input to VTA GABA neurons [26]. Indeed, based on intracellular recordings in vivo, VTA GABA neuron activity is always preceded by a depolarizing current that is sensitive to NMDAR antagonists, which abolishes their spiking. Thus, the activity of VTA GABA neurons appears to be driven strongly by GLUergic synaptic input, even at the high firing rates characteristic of these neurons in vivo.
Based on in vivo findings, we postulated that NIC was acting on α7 nAChRs, to enhance GLU release on VTA GABA neurons. However, the synaptic mechanism of action of NIC on GLU synaptic transmission is best determined in vitro. Thus, we tested the effects of NIC and α7 nAChR agonists on spontaneous and evoked EPSCs in horizontal slice preparation. Nicotine did not alter sEPSC amplitude or frequency, perhaps because of complex temporospatial effects on multiple nAChRs serving VTA GABA neurons. Although choline increased sEPSC frequency in VTA GABA neurons in the slice preparation, this approach inherently possesses two limitations. First, the excitatory GLU input from the PFC that contributes significantly to VTA GABA neuron firing rate [26], and is modulated by α7 nAChRs, is likely severed in the slice, even when cut horizontally. Thus, the action potential-dependent events that compose sEPSCs may not represent PFC GLU input. Second, sEPSCs are likely composed of local-circuit GLU input to VTA GABA neurons [45], as well as action potentialindependent spontaneous GLU release. While GLU input from the PFC may not be spontaneously active on VTA GABA neurons in the slice preparation, action potential-independent spontaneous release of GLU would still be operational, albeit somewhat diluted by other GLU inputs that appear to be local circuit in origin. However, the α7 agonist JN403 increased eEPSC amplitude, providing further support that activation of α7 nAChRs on GLU terminals underlies NIC activation of VTA GABA neuron firing rate.
It is to be noted that the choline used in some in vivo experiments not only has the ability to activate α7 nAChRs, but also α3β4 nAChRs. Nevertheless, we are confident that the NIC actions in these experiments are mediated through α7 nAChRs, as use of the α7 selective partial agonist JN403 induced similar results. In addition, NIC modulation of VTA GABA neurons can be abolished by the α7 selective nAChR antagonist MLA, but not by the α3β4 nAChR sensitive antagonist MEC in vivo. Thus far, there is no evidence of α3β4 nAChR expression in VTA GABA neurons. However, when choline was utilized in VTA slice preparations, the effect of choline on VTA GABA neuron firing cannot be excluded from choline acting on α3β4 nAChRs in other brain areas or neuronal subtypes. This may indirectly affect VTA GABA neuronal function.
Dissociated neuron experiments showed very low percentages of VTA GABA neurons that responded to NIC agonists, although some responses are likely mediated through α7 nAChRs. We assert that these α7 nAChRs on postsynaptic somatodendritic sites may not play an important role in mediating the NIC-induced increase in VTA GABA neuron firing. First, the density of these somato α7 nAChRs appears to be very low. Second, these receptors, even though they may depolarize membrane potential, they desensitize very quickly at high NIC concentrations. Finally, inhibition of ionotropic GLU receptors, such as produced by a NMDAR antagonist, can completely abolish this nicotinic effect.
Finally, to validate the physiological evidence supporting our hypothesis that NIC activation of VTA GABA neurons is through activation of α7 nAChRs on GLU terminals and not on VTA GABA neurons, we examined the expression of nAChR subunits in VTA GABA neurons from GAD GFP mice. A high percentage of VTA GABA neurons expressed α4β2, but only a small percentage expressed α6, and none of them expressed α7 nAChRs, providing molecular evidence in support of our physiological findings.
Our data suggest that α7 nAChRs are mainly expressed at GLUergic terminals onto GABA neurons, which likely play a key role in the mediation of local NIC-induced firing increase in these neurons. The combination of our in vivo and in vitro results, as well as the behavioral data of others, forms a strong justification for the previous affirmation. The results obtained in these experiments further illuminate the functional role of α7 nAChRs in the VTA. Since the experiments from this study were performed to determine the acute effect of NIC on VTA neurons, no direct conclusion can be obtained regarding the addictive activity of NIC after prolonged (chronic) use. Nevertheless, the obtained results elucidate the functional role of α7 nAChRs in GLUergic neurons, supporting the notion that ligands that modulate this nAChR subtype might be important for the development of therapies for nicotine addiction.
Work toward this project was supported by a Brigham Young University Mentoring Grant, and by NIH grant AA020919 to SCS. The authors thank Dr. Dominik Feuerbach (Novartis Institutes for Biomedical Research, Switzerland) for the gift of JN403.
Conceived and designed the experiments: Devin H Taylor, Jie Wu, Hugo R Arias, and Scott C Steffensen; Performed the experiments: Devin H Taylor, Poromendro N Burman, Micah D Hansen, Rebecca S Wilcox, Brett R Larsen, JKB and Collin B Merrill; Analyzed the data: Devin H Taylor, Jie Wu, Collin B Merrill, Jeffrey G Edwards and Sterling N Sudweeks; Wrote the paper: Devin H Taylor, Jeffrey G Edwards, Sterling N Sudweeks, Jie Wu, Hugo R Arias and Scott C Steffensen.