Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2015) Volume 4, Issue 5
Sesamin, a major lignan found in sesame oil, is widely used in traditional Chinese medicine for its bioactivities. However, the information on the neuroprotective effects of sesamin against ischemia- or glutamate-induced excitotoxic injury is limited. This study aimed to investigate the neuroprotective effects of sesamin against focal cerebral ischemia in vivo and N-methyl-d-aspartate (NMDA)-induced neurotoxicity in vitro. Sesamin (43.2 mg/kg) attenuated cerebral ischemic injury in mice induced by 2 h of middle cerebral artery occlusion and reperfusion. Furthermore, treatment with 0.1 μM sesamin significantly decreased the number of apoptotic neuronal cells in cultured neurons after exposure to 200 μM NMDA. Western blot and calcium imaging results indicated that sesamin protected neurons against excitotoxicity by restoring the balance of apoptotic proteins and inhibiting calcium overload in cultured neurons after exposure to NMDA. Our findings provide a new insight into the development of natural anti-excitotoxicity agents.
<Keywords: Sesamin; Apoptosis; Neuroprotection; N-methyl-Daspartate; Calcium
Glutamate is the major excitatory neurotransmitter in the central nervous system (CNS) [1]. However, excessive glutamate is associated with numerous neurological disorders including stroke, traumatic brain injury [2,3], multiple sclerosis, Huntington’s disease, Parkinson’s disease and Alzheimer’s disease [4]. Excessive glutamate can over activate glutamate receptors and result in high calcium ion (Ca2+) influx, which activates a number of enzymes that damage cell structures such as the cell membrane, cytoskeleton components and DNA. This Ca2+ influx is thought to contribute to Ca2+- mediated excitotoxic neuronal cell death in the processes of the abovementioned diseases [5]. N-methyl-D-aspartate (NMDA) receptors, a type of ionotropic glutamate receptors, perform a crucial function in mediating glutamate excitotoxicity because of its high calcium permeability [6]. NMDA receptors are permeable to Na+, K+, and Ca2+ ions, among which excess Ca2+ ions is linearly correlated with NMDA-mediated neuronal cell death triggered by intracellular Ca2+-dependent cascades [7].
Apoptosis is a process of programmed cell death, which is a cascade produced by several apoptosis-regulatory genes [8]. Caspase-3, a member of the caspase family, determines the incidence of apoptosis. The members of the Bcl-2 family such as Bcl-2 and Bax proteins are the most prominent factors in the mitochondria-mediated apoptotic pathway [3,9]. Studies have shown the powerful properties of various natural polyphenols against neuronal exitotoxicity induced by NMDA [10,11]. Sesamin (a typical lignan with β-linkage) and sesamolin (a compound with a phenyl group with an acetaloxygen bridge) are two major lignans in sesame seeds and oil (Figure 1A), which are also found in several medicinal herbs, such as Acanthopanax senticosus. Sesamin is recognized to have various pharmacological effects, such as hypocholesterolemia, anti-hypertension, anti-inflammation, and protective effects against neurotoxicity, hypoxic and oxidative damage [12-15]. The neuroprotective effects of sesamin have been investigated in models of MCAO [15,16]. However, the underlying neuroprotective mechanisms of sesamin are not well known.
The present study was designed to investigate whether sesamin has neuroprotective effects against focal cerebral ischemia in vivo and NMDA -induced neurotoxicity in vitro. The treatment with sesamin was found to produce significant protection against the excitotoxicity triggered by NMDA and against a cerebral ischemic injury induced by a 2 h MCAO and reperfusion.
Chemicals and reagents
Sesamin (purity >98 %) was purchased from Shanghai Pure One Biotechnology (Shanghai, China). NMDA, Glycine, poly- L-lysine, propidiumiodide (PI), 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazoliumbromide (MTT), 2’-(4-Hydroxyphenyl) -5-(4-methyl-1-piperazinyl)-2,5’-bi-1H-benzimidazde, trihydrochloride (Hoechst33258), and β-actin antibody were purchased from Sigma (St. Louis, MO, USA). Anti-Bax, anti-Bcl-2, and anti-pro-caspase-3 antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA). LDH Cytotoxicity Assay Kit was purchased from Beyotime (Beyotime, China). All secondary antibodies conjugated with horseradish peroxidase (HRP) were purchased from Santa Cruz (Santa Cruz, CA, USA). All of the other chemicals and reagents were standard.
Middle cerebral artery occlusion (MCAO).
Experiments were conducted in accordance with the Animal Care and Use Committee of the Fourth Military Medical University. C57BL/6J male mice (20 ~ 25 g) were provided by the Experimental Animal Center of the Fourth Military Medical University. They were housed with food and water available ad libitum in colony room at controlled temperature (25 ± 2ºC), humidity (45 ~ 55 %) and 12:12 h light–dark cycle. To investigate the neuroprotective effects of sesamin on cerebral ischemia, MCAO mouse model was employed in this study [17]. In brief, mice were anesthetized with chloral hydrate (300 mg/kg) and a longitudinal incision of 10 ± 2 mm was made along the midline of the ventral cervical part. The right common carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were exposed and carefully isolated. A nylon mono filament (20 mm length and 0.2 mm diameter) was inserted from the lumen of the ECA to that of the right ICA to occlude the origin of the right middle cerebral artery (MCA). After 120 min, the nylon monofilament was removed to restore blood flow. Temperature was maintained at 37 ± 0.5ºC throughout the surgery. All surgical procedures were performed under an operating stereomicroscope. Mice were randomly divided into three groups: (1) sham operated group, (2) MCAO group (120 min of MCAO followed by 24 h of reperfusion), and (3) sesamin+ MCAO group. The mice in sham operated group were subjected to surgery and exposed the right ICA and the right ECA but did not suffer MCAO. The mice of MCAO and sesamin + MCAO groups were treated with saline or sesamin (43.2 mg/kg) (16) respectively by intragastric administration. Saline or drugs was fed once daily for seven consecutive days before the experiment. MCAO was carried out 30 min after the last drug administration.
Neurological Scoring and Infarct Size Measurement
The neurobehavioral evaluation and infarct volume assessment were performed 24 h after reperfusion in each group. Neurological scores were assessed on a scale: 0, no neurological deficit; 1, failure to extend left forepaw fully; 2, circling to the left; 3, inability to bear weight on the left; 4, no spontaneous walking with depressed level of consciousness [18].
After neurological evaluation, half of the mice (n=6) were killed and the brains were removed and cooled in iced saline for 10 min. Infarct volumes were measured as described previously [19]. Briefly, brains were cut into 1-mm-thick coronal sections and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) for 30 min at 37ºC followed by overnight immersion in 4% formalin. The infarct tissue remained unstained (white) and normal tissue was stained (red). The brain slices were photographed using a digital camera. White areas were defined as infarct and were measured using image analysis software (Adobe Photoshop CS3 for Windows). The infarct volume was calculated by measuring the unstained area in each slice, multiplying it by slice thickness (1 mm), and then summing all six slices.
Nissl staining
After neurological evaluation, another half of mice (n=6) were perfused with cold 4% paraformaldehyde in 0.01 M PBS (pH 7.4). Brain containing dorsal hippocampal and the prefrontal cortex area were cut into frozen coronal sections (30 μm) by using a Leica CM1950 and then stained with 0.1% cresylviolet for 40 min. The images were captured with light microscope (Olympus BX60). Six sections from each animal were selected for Nissl staining. Data were expressed according to the formula: intact neurons (%)=number of healthy neurons/number of total neurons × 100.
Primary mouse cortical neuronal culture
Embryos (E16 ~ 17, C57BL/6J mice for both genders) were employed in this experiment. The Animal Care and Use Committee of the Fourth Military Medical University approved all of the animal protocols used. Prefrontal cortex neurons were cultured as described previously [20]. Briefly, the prefrontal cortex was dissected, minced, and trypsinized at 37ºC for 10 min using 0.25% trypsin (Invitrogen, Carlsbad, CA). Neurons seeded at a density of 2 × 104 cells/well in 96- well plate, 2 × 105 cells/well in 24-well plate containing glass coverslips (Fisher Scientific) and 1 × 106 cells/well in 6-well plate respectively for different treatments. All plates were pre-coated with 50 μg/ml poly-Llysine (Sigma) in water. The cultures were incubated at 37ºC in 95% air/5% carbon dioxide with 95% humidity. The cells were characterized by immunohistochemistry staining for anti-MAP2 antibody, revealing that this culture procedure yielded more than 95% neurons. Cultures were used for experiments on the day 7 in vitro (DIV 7). The culture medium was removed, and cells were washed with Mg2+-free extracellular solution (ECS) containing (in mM): NaCl: 140, KCl: 3, CaCl: 22, HEPES: 10, glucose: 10, adjusted to pH 7.2 ~ 7.3 with NaOH and osmotic pressure 290 ± 5 with sucrose. Then the cortex neurons were exposed for 60 min to ECS supplemented with 200 μM NMDA (Sigma, St. Louis, MO, USA) and 20 μM glycine (Sigma). The cells were washed three times and returned to the original culture medium for 24 h. Sesamin was added into culture medium 24 h prior to addition of NMDA and were present throughout whole experiment.
Cell viability analysis
Neuronal cell viability was determined by MTT assay as previously described [21] with some modifications. Briefly, neurons were used on day 7 (DIV 7). MTT was dissolved in neurobasal medium and added to each well for incubation at 37ºC for 4 h at a final concentration of 0.5 mg/ml. Then the medium was replaced by 150 μl dimethyl sulfoxide (DMSO). The optical density (OD) was read on a Universal Microplate Reader (Elx800, Bio-TEK instruments Inc., USA) at 570 nm (630 nm as a reference). Cell viability was expressed as a percentage of control value.
LDH assay
The LDH activity assay was performed with the LDH Cytotoxicity Assay Kit (Beyotime, China). LDH, a stable cytosolic enzyme, is released upon cell lysis. It was measured with a 30 min coupled enzymatic assay in culture supernatants, which resulted in the conversion of a tetrazolium salt into a red formazan product. The amount of color formed was proportional to the degree of damage to the cell membranes [22]. Absorbance data was collected using a Universal Microplate Reader (Elx800, Bio-TEK instruments Inc., USA) at 490 nm. LDH leakage was expressed according to the formula: LDH release (%)=(experimental LDH release-blank LDH release)/(maximum LDH release-blank LDH release) × 100.
Hoechst/PI double staining
Cell death was detected by Hoechst 33258 and PI double fluorescent staining as described previously [23] with some modifications. Neurons were cultured onto the cover slides in the 24-well plates at a density of 2 × 105 cells/well. Neurons were pretreated with sesamin (0.1 μM) for 24 h. Subsequently, they were subjected to excitotoxic injury with 200 μM NMDA and 20 μM glycine for 60 min. After that, the neurons were returned to the original culture medium including sesamin for 24 h. The cells were washed with ECS for three times, stained with PI (10 μg/ml) and Hoechst 33258 (10 μg/ml) for 15 min, and then fixed by 4 % paraformaldehyde for 10 min. Cells were observed under a fluorescence microscope (Olympus BX61, Japan). The Hoechst and PI dye were excited at 340 and 620 nm respectively. For each well, three visual fields were selected randomly.
Calcium imaging
Calcium imaging was performed as previously described [24,25]. Neurons were cultured in 3.5 cm plates at density 3 × 105 cells. The plate is made especially for laser scanning microscope. Cultured neurons were used on day 9, and then were washed twice using Mg2+-free extracellular solution (ECS). The neurons were incubated with 2.5 μM fluo-3/AM at 37ºC for 30 min, then washed twice and returned to the original culture medium for additional 20 min. Prior to NMDA treatment, the dye-loaded cells were scanned for 1 min to obtain a basal level of intracellular Ca2+ image by using a confocal laser scanning microscope (Olympus, Japan). NMDA (200 μM) was applied to the cultures, and an equal amount of ECS was added as a control. Sesamin was added to the culture medium for 24 h before the NMDA treatments and presented in the whole experiment process. The change of Ca2+ concentration was measured by the fluorescence ratio of the fluo-3/AM loaded neurons for another 4 min. The results expressed as the changes compared to the basal level
Western blot
Western blot analysis was performed as previously described [26]. Protein samples (30 μg) were separated by 9 % SDS-polyacrylamide gel and electrotransferred onto PVDF membranes (Invitrogen). The membranes were blocked by 5% fat-free milk at room temperature for 2 h and incubated with anti-Bcl-2 antibody (1:1000), anti-Bax antibody (1:1000), anti-pro-caspase-3 antibody (1:500), and with β-actin (1:10000) at 4ºC for overnight. After three washes with PBST, membranes were further incubated with HRP-conjugated secondary antibodies for 1 ~ 2 h and followed by three PBST washes. The target protein bands were visualized by an ECL chemiluminescence system (Bio-Rad, Hercules, CA).
Statistical analysis
Results were expressed as the mean ± SEM. Data were evaluated using independent samples test between two groups or one-way analysis of variance (ANOVA) for post hoc comparisons among multiple groups (SPSS 13.0). Data that passed the homogeneity test were analyzed by the one-way ANOVA Least Significant Difference (LSD) test. Data that did not pass the homogeneity test were analyzed by the one-way ANOVA Dunnett’s T3 test. In all cases, p<0.05 was considered statistically significant.
Attenuation of ischemia injury by sesamin
The neuroprotective effect of sesamin against ischemiareperfusion injury was evaluated via neurological scoring, infarct size measurement, and Nissl staining. MCAO resulted in large infarct size (F (2, 15) =360.6, P<0.01, one-way ANOVA, Dunnett T3 test; Figure 1B and 1C) and high neurological deficit score (F (2, 15) =92.5, P<0.01, one-way ANOVA, Dunnett T3 test; Figure 1D). Sesamin (43.2 mg/kg) significantly decreased the infarct volume (F (2, 15) =360.6, P<0.01, one-way ANOVA, Dunnett T3 test; Figure 1B and 1C) and neurological deficit score (F (2, 15) =92.5, P<0.01, one-way ANOVA, Dunnett T3 test; Figure 1A and 1D) as compared with those of MCAO group. The Nissl staining showed that neuronal damage was obvious in the hippocampus CA1 region and prefrontal cortex after MCAO (Figure 2A). The injured neurons showed shrunken cell bodies accompanied by shrunken and pyknotic nuclei. As shown in Figure 2B and 2C, the number of healthy neurons was reduced to 12.6 ± 1.0% in the hippocampus (F (2, 9) =908.3, P<0.01, one-way ANOVA, LSD test; Figure 2B) and 9.3 ± 0.8% in the prefrontal cortex (F (2, 9) =427.8, P<0.01, one-way ANOVA, LSD test; Figure 2C) in MCAO group, respectively. However, sesamin (43.2 mg/kg) markedly rescued the damaged neurons (hippocampus: F (2, 9) =908.3, P<0.01, one-way ANOVA, LSD test; prefrontal cortex: F (2, 9) =427.8, P<0.01, one-way ANOVA, LSD test; Figure 2B and 2C).
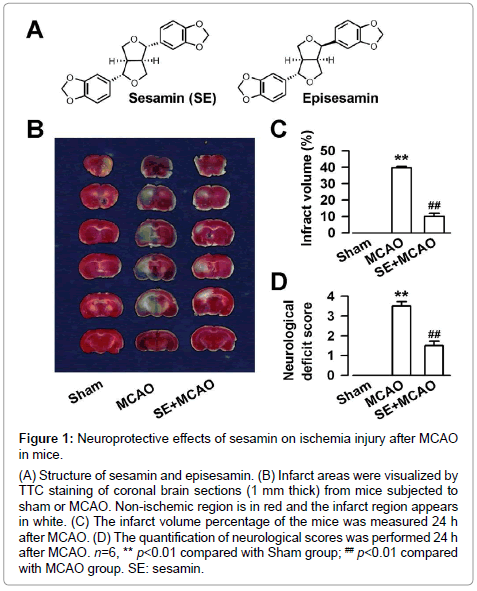
Figure 1: INeuroprotective effects of sesamin on ischemia injury after MCAO in mice.
A) Structure of sesamin and episesamin. (B) Infarct areas were visualized by TTC staining of coronal brain sections (1 mm thick) from mice subjected to sham or MCAO. Non-ischemic region is in red and the infarct region appears in white. (C) The infarct volume percentage of the mice was measured 24 h after MCAO. (D) The quantification of neurological scores was performed 24 h after MCAO. n=6, ** p<0.01 compared with Sham group; ## p<0.01 compared with MCAO group. SE: sesamin.
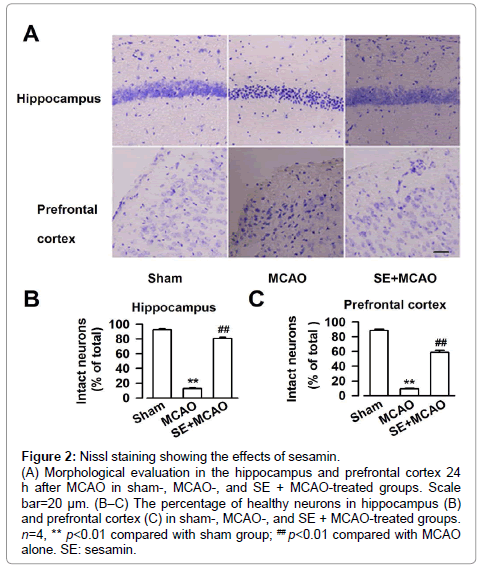
Figure 2: Nissl staining showing the effects of sesamin.
(A) Morphological evaluation in the hippocampus and prefrontal cortex 24 h after MCAO in sham-, MCAO-, and SE + MCAO-treated groups. Scale bar=20 μm. (B–C) The percentage of healthy neurons in hippocampus (B) and prefrontal cortex (C) in sham-, MCAO-, and SE + MCAO-treated groups. n=4, ** p<0.01 compared with sham group; ## p<0.01 compared with MCAO alone. SE: sesamin.
Sesamin protected neurons against NMDA-induced cell death.
To further evaluate the neuroprotective effects of sesamin, primary cortical neuron culture was performed and the neuroprotection of sesamin was detected by MTT assay in vitro. Cultured cortical neurons (DIV 7) were pretreated with sesamin for 24 h and then treated with NMDA for 60 min. The cells were returned to the original culture medium, which contains sesamin, for another 24 h (Figure 3A, top). Exposure to NMDA (200 μM) for 60 min markedly induced cell injury (F (6, 35) =11.6, P<0.01, one-way ANOVA, LSD test; Figure 3A, bottom). Treatment with sesamin (0.1-1 μM) significantly protected neurons against NMDA injury (F (6, 35) =11.6, P<0.01 and P<0.05, one-way ANOVA, LSD test). However, the higher concentration of sesamin (10 μM) failed to protect the neurons against NMDA exposure. Sesamin (0.1 μM) alone did not affect cell viability.
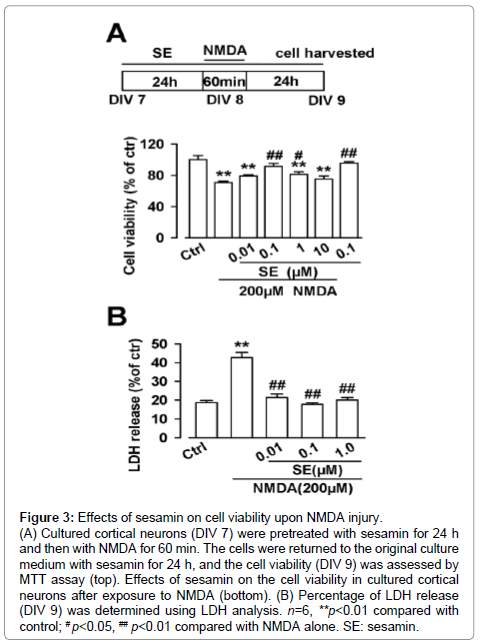
Figure 3: NEffects of sesamin on cell viability upon NMDA injury. (A) Cultured cortical neurons (DIV 7) were pretreated with sesamin for 24 h and then with NMDA for 60 min. The cells were returned to the original culture medium with sesamin for 24 h, and the cell viability (DIV 9) was assessed by MTT assay (top). Effects of sesamin on the cell viability in cultured cortical neurons after exposure to NMDA (bottom). (B) Percentage of LDH release (DIV 9) was determined using LDH analysis. n=6, **p<0.01 compared with control; # p<0.05, ## p<0.01 compared with NMDA alone. SE: sesamin.
To further demonstrate the neuroprotection of sesamin, the amount of LDH release was measured using the LDH assay. NMDA caused a significant amount of LDH release (F (4, 25) =38.8, P<0.01, one-way ANOVA, Dunnett T3 test; Figure 3B), and sesamin (0.1 μM) completely protected the cells from cell damage (F(4, 25) = 38.8, P < 0.01, one-way ANOVA, Dunnett T3 test; Figure 3B). The results were consistent with the data from MTT assay.
Hoechst 33258 and PI double staining were carried out to further determine the neuroprotective effects of sesamin. The results suggested that 8.1 ± 1.4 % cells in the control group and 43.4 ± 4.7 % cells in NMDA group underwent cell apoptosis or death (F (2, 9) =32.3, P<0.01, one-way ANOVA, LSD test; Figure4A and 4B). However, 0.1 μM sesamin markedly attenuated the excitotoxicity induced by NMDA in cultured cortical neurons (F (2, 9) =32.3, P<0.01, one-way ANOVA, LSD test; Figure4A and 4B).
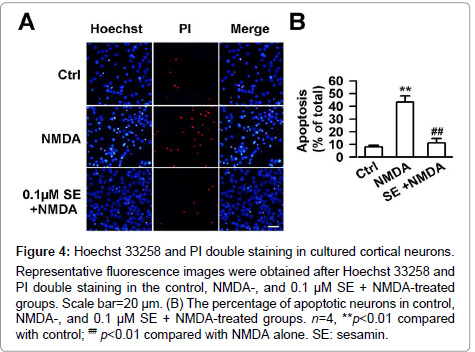
Figure 4: Hoechst 33258 and PI double staining in cultured cortical neurons. Representative fluorescence images were obtained after Hoechst 33258 and PI double staining in the control, NMDA-, and 0.1 μM SE + NMDA-treated groups. Scale bar=20 μm. (B) The percentage of apoptotic neurons in control, NMDA-, and 0.1 μM SE + NMDA-treated groups. n=4, **p<0.01 compared with control; ## p<0.01 compared with NMDA alone. SE: sesamin.
Sesamin reduced the NMDA-evoked Ca2+ increase in cortical neurons
The calcium concentration in the control neuron was stable as shown by the unchanged fluorescence intensity during the whole detection process (Figure 5A and 5B). NMDA (200 μM) treatment produced a persistent elevation of Ca2+ level in cultured neurons (Figure 5A and 5C). However, treatment with sesamin (0.1 μM) reduced the NMDA-evoked increase of Ca2+ concentration as shown by decreased fluorescence intensity (T=8.0, p<0.01, independent samples test; Figure 5A and 5C).
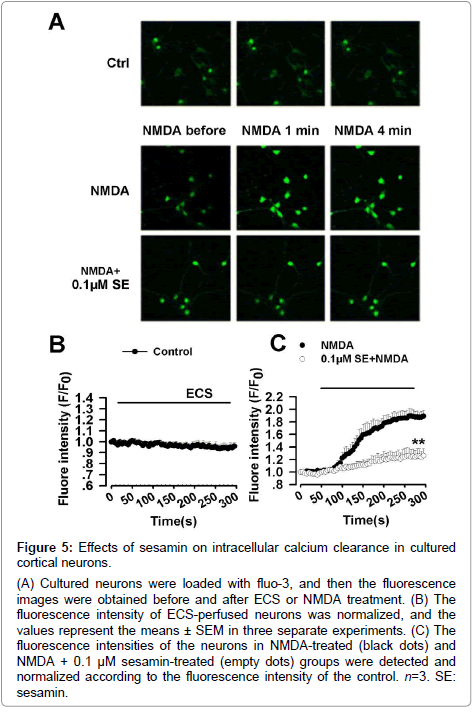
Figure 5: Effects of sesamin on intracellular calcium clearance in cultured cortical neurons.
(A) Cultured neurons were loaded with fluo-3, and then the fluorescence images were obtained before and after ECS or NMDA treatment. (B) The fluorescence intensity of ECS-perfused neurons was normalized, and the values represent the means ± SEM in three separate experiments. (C) The fluorescence intensities of the neurons in NMDA-treated (black dots) and NMDA + 0.1 μM sesamin-treated (empty dots) groups were detected and normalized according to the fluorescence intensity of the control. n=3. SE: sesamin.
Effects of sesamin on the expression of apoptosis-related proteins
Whether the protection of sesamin was associated with the changes in the expression of apoptosis-related proteins was determined. As shown in Figure 6, NMDA treatment down-regulated the Bcl-2 expression (F (2, 9) =17.0, P<0.01, one-way ANOVA, LSD test; Figure 6A and 6B), up-regulated the Bax expression (F (2, 9) =28.5, P<0.01, one-way ANOVA, LSD test; Figure 6A and 6C), and thus increased the Bax/Bcl-2 ratio (F (2, 9) =124.9, P<0.01, one-way ANOVA, LSD test; Figure 6D). However, 0.1 μM sesamin showed anti-apoptotic activity with increasing Bcl-2 expression (F (2, 9) =17.0, P<0.01, one-way ANOVA, LSD test; Figure 6A and 6B), decreasing Bax expression (F (2, 9) =28.5, P<0.01, one-way ANOVA, LSD test; Figure 6A and 6C), and decreasing Bax/Bcl-2 ratio (F (2, 9) =124.9, P<0.01, one-way ANOVA, LSD test; Figure 6D). Caspase-3 is known to have a crucial function in the apoptotic process. Pro-caspase-3 is an inactive precursor of caspase-3 and cleaves into active caspase-3, thereby causing Bcl-2 dialysis [27]. Subsequently, changes in the pro-caspase-3 expression were observed. Cultures treated with NMDA resulted in the decrease of pro-caspase-3 (F (2, 9) =12.1, P<0.01, one-way ANOVA, LSD test; Figure 6A and 6E). Sesamin (0.1 μM) significantly increased the levels of pro-caspase-3 (F (2, 9) =12.1, P<0.05, one-way ANOVA, LSD test; Figure 6A and 6E).
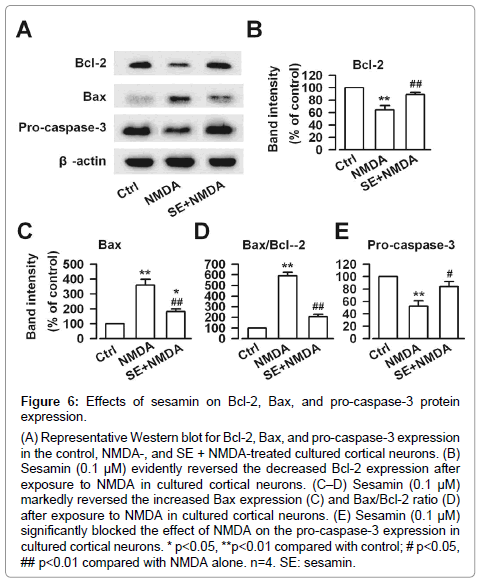
Figure 6: Effects of sesamin on Bcl-2, Bax, and pro-caspase-3 protein expression.
(A) Representative Western blot for Bcl-2, Bax, and pro-caspase-3 expression in the control, NMDA-, and SE + NMDA-treated cultured cortical neurons. (B) Sesamin (0.1 μM) evidently reversed the decreased Bcl-2 expression after exposure to NMDA in cultured cortical neurons. (C–D) Sesamin (0.1 μM) markedly reversed the increased Bax expression (C) and Bax/Bcl-2 ratio (D) after exposure to NMDA in cultured cortical neurons. (E) Sesamin (0.1 μM) significantly blocked the effect of NMDA on the pro-caspase-3 expression in cultured cortical neurons. * p<0.05, **p<0.01 compared with control; # p<0.05, ## p<0.01 compared with NMDA alone. n=4. SE: sesamin.
Sesamin produces effective neuroprotection against cerebral ischemia [15]. Sesamin has been reported to rescue neuronal PC12 cells from apoptosis and death induced by the MPP+ activation of microglia cells [28], protect neuronal PC12 cells from high-glucose-induced oxidation and apoptosis [29], and protect neurons against hypoxic neuronal injury [30]. Sesamin is also a free radical scavenger, which has been proven to exhibit prominent antioxidant and free radical scavenging properties [31-33]. In the present study, a mouse model of transient focal cerebral ischemia induced by a 2 h MCAO and 24 h reperfusion was used to evaluate the neuroprotective effects of sesamin. Previous studies have reported that MCAO damages the structure of the neurons and other cells that support the neurons [20,34] and decreases neurological function [20]. Our results suggested that sesamin reduces the infract volume in the brain of MCAO mice and improves the neurological deficit score. Meanwhile, the abnormal neuronal morphology in prefrontal cortex and hippocampus is ameliorated.
Glutamate is the main excitatory neurotransmitter in the CNS. High glutamate concentration induces excitotoxicity, which injures neuronal cells under in vitro and in vivo conditions [35]. NMDA receptor is a type of ionotropic glutamate receptor that regulates cell survival in vivo as well as in vitro. Over-stimulation of NMDA receptors leads to neuronal excitotoxicity because the subsequent influx of free Ca2+ results in the overload of intracellular Ca2+, which has a vital function in NMDA-induced neuronal cell apoptosis [36]. Calcium influx activates a number of enzymes that damage cell structures such as the membrane, components of the cytoskeleton, and DNA. Many studies have shown that Ca2+ overload triggers several downstream lethal reactions, including cGMP elevation, NOS activation, and glutamate release [37,38]. In this study, the results of the MTT assay, LDH assay, Hoechst 33258 and PI double staining, and Fluo-3/AM fluorescent intensity test show that NMDA induces neuronal injury, which is consistent with previous reports [9,22]. Sesamin reduces neuronal apoptosis and inhibits the elevation of intracellular Ca2+ triggered by NMDA. These effects may be involved in the neuroprotection of sesamin against excitotoxicity.
A number of proteins influence and guide the apoptotic progression of mitochondrial pathway. The family of Bcl-2 proteins contains both the pro-apoptotic molecule Bax and the anti-apoptotic molecules Bcl- 2 and Bcl-XL [39]. Their functions on cell survival in various stimuli including cytotoxic injury and oxidative stress have been well studied. The balance between Bcl-2 and Bax plays critical roles in regulating of cell death or survival [40]. The increase in the Bax/Bcl-2 protein ratio has been shown to be responsible for excitotoxic apoptosis and death mediated by NMDA in neurons [41]. The activations of initiator caspase-9 and the effector caspase-3 are very important in the apoptosis pathway. Caspase-3 activation is associated with stroke-induced apoptotic neuronal cell death [42,43]. Pro-caspase-3 is an inactive form of active caspase-3 and a major physiologic target of caspase-8 and -9 [44]. Our study showed that sesamin (0.1 μM) significantly increases the expression of Bcl-2 protein, decreases the Bax expression, and restores the increasing ratio of Bax/Bcl-2 induced by NMDA. The high level of pro-caspase-3 is related with high survival rate in cells [45]. The level of pro-caspase-3 is obviously decreased after NMDA exposure in cultured neurons, suggesting the activation of pro-caspase-3 to caspase-3. However, sesamin (0.1 μM) markedly restores the level of pro-caspase-3. A study demonstrates that sesamin can diminish iNOS and COX-2 protein expressions and significantly restore the SOD activity and protein expression levels in the acute hepatic injury rats [46]. Furthermore, sesamin treatment also significantly reduces inflammatory and oxidative stress markers including Iba1, Nox-2, and Cox-2 [15].
There are limitations of the present study. First, different protocols were used for the in vivo and in vitro experiments. Sesamin was used for seven days before the MCAO in vivo experiment, which is the standard protocol for in vivo studies. For the in vitro study, the cells were exposed to sesamin both before and after NMDA administration. The concentration of the drugs (sesamin in the present study) was speculated to decrease gradually in vivo. Actually, the pharmacokinetics of sesamin was not detected. Therefore, whether the pretreatment or post-treatment with sesamin was associated with neuroprotection could not be discriminated in the present study. More experiments need to be performed to reveal the best time window for sesamin treatment. Furthermore, there are a sex-dependent response to excitotoxicity, oxygen-glucose deprivation and nutrient deprivation in neurons in vitro [47]. Therefore, mixing male and female neurons in the present cultures may not be adequate, as differences in the ratio between male/female neurons could result in a different response to the treatments.
In summary, the current study investigated the neuroprotective effects of sesamin on focal cerebral ischemia in vivo and against NMDA-induced cell apoptosis in vitro. This effect is partially attributed to anti-apoptotic activities. However, more experiments are necessary to elucidate the underlying mechanisms for the neuroprotective effect of sesamin.
This work was supported by NSF of China No.31470052, 31271126, and the Program for Elite Talents in University 4138C4IA75.
The authors indicated no potential conflicts of interest