Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Special Issue Article - (2013) Volume 0, Issue 0
Human polyglutamine (poly (Q)) induced neurodegenerative disorders are fatal illnesses characterized by progressive loss of neurons in specific areas of brain resulting to various neurological complications leading to death. The disease manifestation is age dependent and major symptoms include abnormal body movement, cognitive deficits, dementia, psychosis, tremors and spasticity. Ageing appears to have a direct impact on poly (Q) disease progression and severity of symptoms is greatly influenced by several intrinsic factors. This review attempts to provide an overview of human poly (Q) diseases and also discuss about the factors affecting progression of neurodegeneration. It also provides an outline of various therapeutic approaches which could be potentially useful to combat these tragic human diseases.
<Keywords: Neurodegeneration, Poly (Q), Protein folding, Ageing, Insulin signalling, Chaperone
Polyglutamine (poly (Q)) diseases comprise a group of neurological disorders exhibiting common phenotypic manifestations and causative factors. However, they could be classified as per their exclusive pattern of brain degeneration. The symptoms are late onset, progressive in nature and cause degeneration of specific population of neurons in brain. Some of the widespread symptoms include abnormal body movement, cognitive deficits, dementia, psychosis, tremors and spasticity. Poly (Q) diseases comprise a set of nine neurological diseases. These are Spinal and bulbar Muscular atrophy (SBMA), Dentatorubral pallidoluysian atrophy, Huntington’s disease (HD) and six kinds of the Spinocerebellar ataxias (SCA 1, 2, 3, 6, 7, 17). Except Spinal and Bulbar Muscular Atrophy (X-linked) all of them are dominantly inherited and affects mostly in adult populations.
The earliest case of poly (Q) disease to be characterized was Huntington’s chorea by George Huntington, in 1872 [1]. It was called chorea due to dance like movement of patient suffering from the disease. For many years the underlying mechanism and cause of the disease remained obscure but the advancement in biological sciences in later part of twentieth century paved way to the understanding of poly (Q) diseases. In 1991, a novel discovery showed that the expansion of CAG repeat in the coding sequence of androgen receptor was the cause of X-linked neuromuscular disorder, commonly known as Spinal and Bulbar Muscular Atrophy or Kennedy’s disease [2]. Subsequently, gene responsible for causing HD was also identified and correlated with expansion of CAG repeats in resident gene [3]. Remaining poly (Q) diseases were also found to be caused by expansion of CAG repeats in various genes. At the same time, another triplet disease known as Fragile X syndrome of Mental retardation (FRAXA) was also unveiled to be developed by extended CGG repeats [4].
The above discoveries made a paradigm shift in the understanding of the pathogenesis of these complex diseases which were once very difficult to decipher. The unusual expansion of CAG repeats provides toxic gain of function and this triggers a large scale degeneration of selected neurons in the brain and spinal cord. Each of the poly (Q) diseases has certain threshold limit of CAG repeats beyond which the disease is manifested at some point of adult life. The threshold length of CAG repeat in HD and ataxia-3 is 36 and 50, respectively. It was found that the extent of CAG repeats above the threshold limit aggravates the disease severity and causes early predisposition of the symptoms.
One of the characteristic features of poly (Q) diseases is genetic anticipation; the subsequent generation of disease affected patient is more likely to inherit longer form of polyglutamine repeats resulting acute severity of the disease [5]. In most of the poly (Q) diseases more of the elongated form of CAG repeat is contributed by paternal side. This is primarily because the expansion of CAG repeats occurs during spermatogenesis and in patients suffering from HD, the testicular cells show high frequencies of repeat expansions [6]. In Dentatorubral- Pallidoluysian Atrophy (DRPLA) also, age of father affects the expansion length of CAG repeat [7]. Therefore, in juvenile form of HD, where the phenotypes of disease appears at very early stage, is most probably contributed by father with highly elongated CAG repeats. The elongation of repeats during spermatogenesis is very mysterious and various studies have been performed to unlock the mechanism. The probable reason has been postulated to be the errors during replication and proofreading. In vivo studies in yeast have shown that repeats affect the replication dependent repair mechanism during spermatogenesis. The elongated CAG repeat leads to formation of hairpin loop and stalls the DNA polymerase. In this situation the DNA polymerase backs up and recopies the opposite daughter strand. The DNA polymerase again restarts to copy the original strand and in this processes elongation of repeats may occur [8]. Even in somatic tissues the repeat expansion and instability is observed and attributed to miss-match repair and base excision repair defects.
The normal copy of the poly (Q) disease causing genes is expressed differentially in various tissues and organs. They perform some vital cellular functions in normal disease free condition. In the brain, Huntingtin expression is ubiquitous and highly concentrated in striatum and cerebral cortex. Moderate level of expression is observed in peripheral tissues such as heart, liver, lungs and testes [9]. The Huntingtin protein is essential for embryonic development and loss of function mutation leads to embryonic lethality in mouse. It has also been suggested to be involved in cellular trafficking and vesicular transport.
Spinocerebellar ataxia type 3 (SCA3) is the most common form of ataxia and is caused by mutation of ataxin 3 located on 14q24.3-32 chromosome. It is also commonly known as Machado-Joseph disease named after the resident brothers of Azores who are considered to be first clinically identified patient suffering from SCA3. Ataxin 3 codes for one of the smallest proteins of 42KDa being expressed in cerebellar cortex, globus pallidus, thoracic spinal cord and lumbar spinal cord. Ataxin 3 is a member of josephin family of proteins which perform the vital function of deubiquitinization. It plays a crucial role in ubiquitinproteasome system and helps to maintain the protein quality control. SBMA is the only X-linked disorder among all the poly (Q) diseases and is causes by mutation in androgen receptor gene. Affected individuals suffer from many complications such as Dysrhythmia, Dysphagia and also Gynocomastia due to androgen insensitivity. DRPLA is caused by mutation of atrophin-1 localizing on 12p13.31. Atrophin-1 is involved in c-Jun N-terminal kinase (JNK) pathway. The expression of atrophin-1 is high in frontal cortex, temporal cortex, basal ganglia and in various part of brain stem [10]. The Ataxins 1, 2, 6, 7 and 17 are mainly associated with regulation of transcription. Their expression is mainly confined to cerebellar cortex, brain stem, spinal cord and basal ganglia.
Although mutant forms of polyglutamine proteins are expressed dynamically, only some selective neurons in the brain are more vulnerable to toxic gain of function. This phenomenon can be attributed to the microenvironment of the neurons which might complement the toxicity. In HD, the spiny neurons of caudate nucleus in basal ganglia are severely degenerated. MRI scans shows atrophy of caudate nucleus, frontal cortex, and thalamus. The post-mortem brain sections of HD patients also show severe degeneration of cortical region, and therefore, a drastic reduction in size [11]. The loss of cognitive ability in Huntington patients may be directly correlated to such changes in brain volume especially the loss of cortical neurons [12]. In Ataxia 3, Purkinje neurons of the cerebellum undergo cell death. In vivo studies on SCA3 show degeneration in the cerebellum, parts of cortex and in pons and basal ganglia. The volume of cerebellum and brain stem are also reduced significantly in both SCA3 and SCA6. The atrophy of cerebellum and brain stem is progressive with respect to age [13]. Initially, the expansion of CAG repeats was thought to be exclusively neurotoxic but mounting evidences suggest that it affects other peripheral tissues as well. In addition to the neuronal dysfunction, HD patients also exhibit various other abnormalities which include dysfunction of cardiac and skeletal muscles and aberrant glucose metabolism [14].
The genetic determinants of the poly (Q) diseases are well known but the actual molecular mechanisms underlying these diseases are poorly understood. A gene with additional CAG repeats in its coding sequence leads elongated glutamine tract in the amino acid chain resulting conformational instability and damage to the native protein structure. The misfolded protein interacts with other proteins containing CAG repeats and interrupts their molecular functions. Another striking feature of polyglutamine proteins is their tendency to form molecular aggregates that are commonly known as Inclusion Bodies (IBs). They form insoluble intracellular aggregates by binding with the molecular chaperones which are essential to combat cellular stresses and for maintenance of cellular homeostasis. The inclusion body formation is a hallmark and the most common characteristic feature of all the poly (Q) diseases [15]. It has been found that the molecular chaperones such as HSP/HSC70 are sequestered in the aggregates and reduces cellular pool of HSP70 [16]. Subsequently, IBs grow in size with the progression of age and get translocated into the nucleus; where they sequester many of the critical transcription factors such as CBP, TAF-II 130, TBP, Sin3 [17]. These sequestrations make a global impact on transcriptional activity which adversely affects the survival of the neurons. One factor that caught major attention of researchers is CBP which is a key regulator of transcription. It has histone acetyltransferase activity and acetylates histones lead to loosening of chromatin core particles, and therefore, making DNA readily available for the action of RNA polymerase and other transcription factors [18]. The sequestration of CBP by IBs leads to hypoacetylation of histones and deflation of transcriptional activities causing deprivation of cellular survival factors and finally apoptosis.
Similar to the polyglutamine neuropathology pattern, investigations in aged human brains suggest reduced expression of the groups of genes which are involved in synaptic function, calcium signalling, vesicular transport, mitochondrial function and protein turnover due to increased DNA damage [13]. This overlapping scenario of, cellular and molecular changes occur during poly (Q) pathology and ageing indicates a recurrent interaction between poly (Q) and ageing pathways. Interestingly, DNA sequence flanking the CAG domain contributes largely to the dynamics of inclusion body formation. In HD, sequence flanking the CAG repeat in first exon seems crucial for inclusion body formation [19]. Similarly in Ataxia 3, a highly conserved domain Josephin is known to play a key role in inclusion body formation.
IBs also have profound effect on axonal transport system of the neurons. They are reported to localize in the axonal routes impairing transport processes. In HD, the IBs localize to the axon terminals resulting in blockage of anterograde and retrograde transport [11,20]. In SCA3, IBs of various sizes localize to axons and sequester many essential transport proteins such as synaptobrevin and glutamate receptors and lead to altered synaptic functions [21].
Despite a number of intriguing pointers, limitations of human genetic studies make it difficult to analyse insights of candidate genes and pathways in greater details, primarily because of complex patterns of inheritance, lack of sufficient family pedigree data and populationbased genetic heterogeneity [22]. Therefore, attempts have been made to model human neurodegenerative diseases in various organisms ranging from unicellular bacteria to rhesus monkey. However, such studies required establishment of model organisms in order to carry out comprehensive analysis on the molecular and cellular events participating in pathogenesis of the disorders.
Since the time TH Morgan first used the small invertebrate, Drosophila melanogaster, to demonstrate the chromosomal theory of inheritance, the scientific community has come a long way in terms of exploiting the powerful genetics offered by fruitfly. Therefore, identification of genes associated with inherited forms of human neurodegenerative diseases during the past decades encouraged the researchers to investigate if Drosophila could be utilized to decipher the underlying mechanisms of disease pathology and to design therapeutic strategies. Drosophila appeared to be a reasonable alternative in this respect owing to the similarity between some of the fundamental cellular processes with humans and the fact that flies and humans share many structurally and functionally related gene families. It is increasingly clear now that approximately 75% of the human disease genes have functional Drosophila homologues [23,24] and the extent of overall identity ranges from 40% between homologues to as much as 80-90% between conserved functional domains [25]. Several other advantages that Drosophila offers over other conventional model systems such as yeast, Caenorhabditis elegans and human cell cultures include its genetic tractability, faster time frame due to a short generation time of approximately 10 days and short life-span of 45-60 days, ease and affordability of maintaining large population within the confines of a laboratory, high fecundity (100 eggs per day per pair), presence of balancer chromosomes, a well worked out developmental processes and anatomy, absence of meiotic recombination in males, relatively small genome contained on four completely sequenced and annotated chromosomes and ability to perform large-scale genetic screens to identify potential modifiers of known disease phenotypes. In addition, availability of a large number of various mutant lines available at several Drosophila stock centres also makes it a popular model organism [26,27].
Furthermore, the adult fly harbours a sophisticated brain and nervous system organized into specialized neural centres capable of exhibiting complex behaviours such as learning and memory, much like the human brain. Disruption of such well-coordinated motor behaviours serves as a highly sensitive readout of neuronal dysfunction and death. Drosophila models also prove to be extremely useful while carrying out pharmacological screens for identifying novel therapeutic drug targets as they lack a blood brain barrier that can prevent the access of the drug to the central nervous system tissues. Moreover, the response towards many drugs that act within the CNS is similar to the effects observed in mammalian systems [25,28].
The dominant nature of most of the mutant alleles of neurodegenerative disorders suggested that perhaps such diseases could be modelled in Drosophila by introducing the mutant allele into the fly genome and producing transgenics. A binary system is used for this purpose which benefits from the property of the yeast transcriptional activator GAL4 protein to bind to a small Upstream Activator Sequence (UAS) of genes to drive transcription. Foreign genes can be cloned into transposable P-element vectors under the control of a UAS sequence and crossed with transgenic flies expressing the yeast GAL4 gene in a tissue-specific manner [29]. At this point it is important to note that the transgene remains silent until crossed with a desired GAL4 line. Most commonly used promoters in case of neurodegenerative disorders include pan-neuronal elav driver and eye specific glass multiple reporter element (gmr) driver. Neurodegeneration can be monitored either by measuring the loss of visible photoreceptors neurons in the eyes or by evaluating motor function by the climbing assay or by checking for the lethality of the organism. There are many advantages of using the UAS-GAL4 system for transgenic expression: first, the conditional nature of expression which prevents deleterious effects of constitutive expression of transgene; second, a single UAS transgenic line can be ectopically expressed in many different tissue types, depending upon the GAL4 line being used; third, the system is capable of yielding much higher levels of expression of the gene of interest as compared to direct promoter-fused transgenes.
One of the most valuable tools that Drosophila bestows for neurodegenerative disease modelling is the use of its compound eyes for easy and direct evaluation of neurodegeneration. Several reasons contribute towards preferring eyes over other organs for genetic studies. These include the ease with which neurodegeneration can be scored in eyes; its well-defined developmental events; and its indispensable nature for the viability of the fly allowing for isolated analysis of genes that might be organismal lethal. Drosophila adult eye is a compound structure made up of a hexagonal array of 800 virtually identical lens-like units called ommatidia. Each ommatidium in turn made up of an ordered arrangement of eight light-sensitive neurons called rhabdomeres (R1-R8) that are surrounded by support cells and pigment cells. Such a highly systematic layout of the eye retina amplifies even minor perturbations in cell patterning that might arise during modifier screens.
Several approaches have been exploited so far to study neurodegeneration in Drosophila. Initially, the classical forward genetic approach was utilized wherein random mutations were created to select a phenotype showing brain degeneration which was then analysed with the aim of identifying the gene responsible for the mutant phenotype [30,31]. However, such screens either remain incomplete due to ignorance of redundant loci and epigenetic effects or are difficult to undertake for certain phenotypes that lack directly measurable phenotypes. Additionally a classical genetics approach takes significantly longer. So, alternative reverse genetics-based approaches have been undertaken to elucidate pathogenic pathways and remedial strategies of neurodegenerative disorders. The major stages include are: i) mis-expression of a human disease gene, in its wild type or mutant form by means of the UAS-GAL4 system; ii) loss/gain of function mutations of the Drosophila homolog of a human disease gene using transgenic over expression or UAS-GAL4-mediated RNAi approaches; iii) modifier screens to identify enhancers or suppressors of a particular disease phenotype and iv) a pharmacological approach to screen for compounds that can potentially prevent or ameliorate the disease phenotype. The efficacy of a compound to act as a potential drug can be evaluated by simply adding a defined concentration of the compound to the usual fly flood and thereafter, feeding the flies on treated vs untreated food and comparing neuronal differentiation and survival in both of the cases.
Majority of Drosophila models for polyglutamine diseases have been developed by employing the misexpression approach discussed above. The first transgenic Drosophila model of a human neurodegenerative disease was described for SCA3. Targeted expression of a C-terminally truncated domain of SCA3 carrying 78 residues of glutamine repeats (SCA3trQ78) induced degenerative phenotypes in Drosophila [32]. This was closely followed by creation of a transgenic model for HD in which exon 1 of the human huntingtin (Htt) protein bearing 2, 75 or 120 glutamine residues respectively were expressed in Drosophila compound eyes [33]. Both the models displayed characteristics of disease phenotypes. Later, Drosophila model for SCA1 using complete full-length protein and spinal and bulbar muscular atrophy (SBMA) models were also generated [34,35]. The SBMA model was produced by expressing human androgen receptor (hAR) protein carrying 52 glutamine residues (hARQ52) into Drosophila. However, unlike other models, these flies failed to exhibit any effect unless supplemented with a ligand for the androgen receptor.
In all the above cases, the expression of the transgene containing an expanded (above threshold level) poly (Q) stretch resulted in the development of retinal degeneration and formation of necrotic patches on Drosophila eye surface. Other phenotypes included depigmentation of the eyes, loss of neuronal integrity and roughing of external eye surface. The pathogenic forms of the alleles were shown to induce a late-onset progressive neurodegenerative phenotype as compared to the normal allele although both the flies were born with normal retinal morphology. For instance, neurodegeneration was observed upon inducing expression of SCAtr(Q)78 while SCAtr(Q)27 did not exhibit any effect. One of the major differences observed with respect to disease pathology in the SCA3 and HD models was that the mutant SCA3 protein led to the formation of nuclear inclusions characteristic of human SCA3 disease while the mutant Htt protein did not participate in nuclear inclusion formation although it exhibited nuclear accumulation. Nevertheless, a link between the nuclear localization of the mutant proteins and the pathogenic events could be established. Furthermore, it was demonstrated that expression of HD pathogenic protein causes defects in axonal transport machinery and culminates in cell death [36]. However, a full-length Htt protein did not display axonal transport blockage; rather, increased neurotransmission seemed to be the cause of early pathogenesis in this case [37]. Severity of the degeneration was found to have a direct correlation with polyglutamine repeat length and the extent of the expression of mutant protein achieved in fly [38]. Pan-neuronal Gal4 (elav-Gal4) driven expression of various disease transgene in Drosophila either causes early lethality, tremors and adult mortality or loss of coordination and late adult mortality [33]. Formation of IBs and sequestration of a large number of vital cellular proteins were also observed in affected tissues. Taken together, Drosophila disease models have been found to successfully recapitulate most of the key features of human neurodegenerative disorders.
Ageing is a major challenge to every living organism characterized by reduced performance in physiological processes and functional capacity of the organism. These physiological changes result in decline of functional efficiency, reduced homeostasis and ultimately, death of the organism. Interestingly, rate of ageing varies between individuals and groups, as well as among different systems, organs and cells within the individuals itself. Physiological changes like increased accumulation of toxic metabolic products and cell damage expose the organism to many kinds of diseases and eventually, they become susceptible to many age dependent diseases where neurodegenerative disorders and cancers pose as major threats [39]. Among the age related disorders, neurodegenerative disorders emerged as an essential area in neurobiology because of disease irreversibility and lack of effective treatment. Recent studies suggest several common cellular and molecular pathways which become dysfunctional in neurodegenerative diseases and ageing, and therefore, indicate their potential association. Thus, in order to ameliorate ageing and age dependent neurodegenerative disorders, understanding the key molecular and cellular pathways that regulate longevity and its correlation to disease pathogenicity become a very basic necessity. Following section of review deals with emerging trends between poly (Q) disorder and ageing.
The past few decades have witnessed an upsurge in the genetic studies on ageing and the elucidation of pathways and mechanisms responsible for the process of longevity. Since a life time as long as that of humans cannot be spent on a single experiment of human ageing, therefore, animal models need to be established. Classical model systems such as C. elegans and Drosophila melanogaster have emerged as excellent organisms to elucidate essential genetic/cellular pathways of human ageing. Over other systems, a few additional advantages presented by the Drosophila for ageing studies included i) a clear distinction between the stages of development and reproduction and ii) presence of post-mitotic cells almost entirely throughout the adult fly, representing synchronised ageing [40]. Elucidating the first aspect, the commencement of adulthood in Drosophila is said to occur only after the fly eclose out of the pupal case. It is during this stage of its life that it is sexually mature and competent to reproduce and thus, ageing is thought to take place now. This is in great contrast with other model systems where it is often difficult to determine when the organism has reached maturity [41]. The second aspect focuses on the rarely dividing neurons of the brain which makes the insect brain an excellent model for the cytological studies of human ageing [42]. A synchronously ageing set of cells can be easily observed for structural changes and conclusive results can be deduced. Moreover, the insect brain lacks the presence of any blood vessels and so, pathological changes pertaining to blood vasculature can be excluded.
The genetic approaches those are commonly applied for ageing studies include mutagenesis followed by forward genetics analysis, molecular genetics, QTL analysis and selective breeding [43]. These methods, so far, have identified several genes that when mutated lead to modulation of lifespan. These genes found to encode a wide variety of proteins essential for diverse cellular functions. Therefore, process of ageing appears to be operated by several cellular aspects.
Despite the expression of toxic protein throughout life, like other neurodegenerative disorders, age emerges to be a critical factor for the onset of poly (Q) diseases. Neuronal loss, shrinkage of cell bodies and axons of neuronal cells and loss of synapse collectively result in reduced brain volume and weight in ageing individuals, who are cognitively normal [44]. Existence of sparse distribution of neurofibrillary tangles and senile plaques which are the neuropathological hallmark of Alzheimer’s disease (AD), have been reported in cortical region of ageing brain that are associated with mild cognitive impairment [45]. Similarly, degeneration of neuronal cell bodies, axons, synapse and specific parts of the nervous system comprise a general pathology of polyglutamine mediated toxicity in various poly (Q) disorders.
Although, it looks quite appealing to hypothesize that disease related proteins might have caused disease toxicity by accelerating the ageing process but it is still unclear whether ageing related changes are responsible for driving neuronal pathology or both ageing and disease associated proteins act synergistically to develop neuronal dysfunctions. In C. elegans, mutation that extends longevity in poly (Q) disease reveals age dependent reduction in protein aggregate formation and decrease in toxicity, consequently testifying the effect of ageing in poly (Q) mediated cellular dysfunction [46]. Recent findings in our laboratory motivate the correlation of ageing in enhancing the poly (Q) mediated neurodegeneration in age dependent manner. Targeted expression of htt-93(Q) in Drosophila eye induced cellular degeneration characterized by retinal depigmentation and cellular toxicity. Our studies on individual flies expressing htt-93(Q) transgene during ageing suggest that the magnitude of retinal depigmentation and cellular toxicity increases with age (Figure 1). Moreover, involvement of common signalling networks in longevity and mitigation of poly (Q) toxicity raise the prospect that slowing down ageing may act as a neuroprotective measure. Therefore, in order to cultivate novel strategies to prevent onset and progression of such deadly disorders, it will be interesting to explore how ageing dysfunction and poly (Q) mediated neuropathology are interlinked and how they interact during disease pathogenesis.
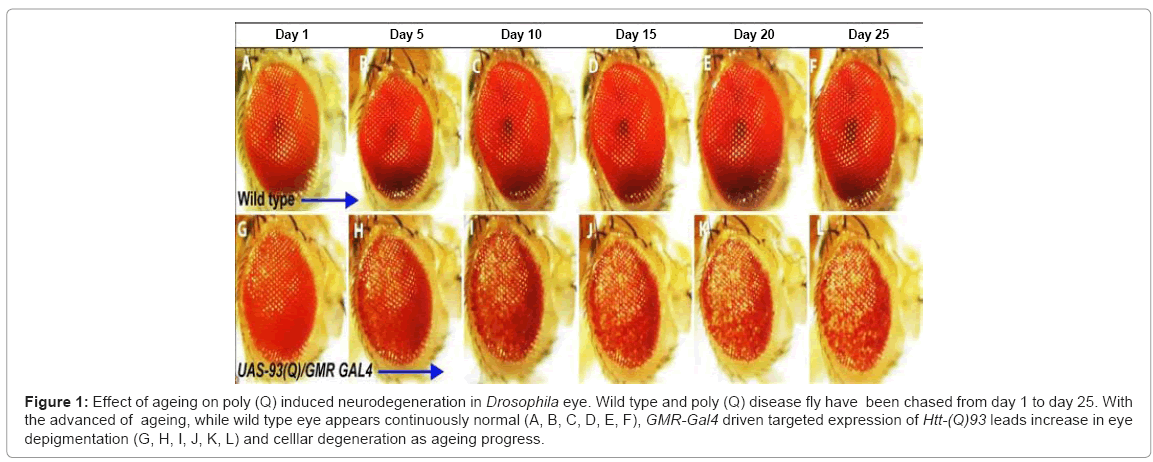
Figure 1: Effect of ageing on poly (Q) induced neurodegeneration in Drosophila eye. Wild type and poly (Q) disease fly have been chased from day 1 to day 25. With the advanced of ageing, while wild type eye appears continuously normal (A, B, C, D, E, F), GMR-Gal4 driven targeted expression of Htt-(Q)93 leads increase in eye depigmentation (G, H, I, J, K, L) and celllar degeneration as ageing progress.
Eukaryotic systems have developed a well regulated protein quality control system which is collectively formed by chaperone network, ubiquitin-proteosome system and lysosome-mediated autophagy. This system is operational during post translational modification, protein folding, stress response and clearance/ translocation of damaged proteins. Molecular chaperones are ubiquitous, highly conserved protein family mainly represented by Heat Shock Proteins (HSPs) those assist in folding and assembly of newly synthesized or damaged misfolded proteins, transportion, degradation etc. [47,48]. These chaperones range from molecular mass of 15 to 110 kDa, and are grouped into 5 major families: HSP100, HSP90, HSP70, HSP60, and the small HSP (sHSP) families. In addition, ubiquitin-proteosome system and lysosome-mediated autophagy drives degradation of misfolded proteins in order to prevent their accumulation in form of toxic aggregates [49].
Ageing is associated with tremendous increase in damaged proteins due to reduced proteosome activity, mitochondrial dysfunction and defect in chaperone network [48,50]. During ageing and disease stress condition, induction and functional capacity of chaperones as well as cellular proteosome activity gets altered; consequently the post mitotic neurons become vulnerable to toxic protein aggregates and ultimately, compromises cell survival. Decades of investigations have suggested involvement of chaperone proteins in ageing and longevity regulation [51]. An increase in chaperone induction has been associated with increased longevity and stress resistance in various animal models including cell line, C. elegans, Drosophila, which further encourage the view of chaperone dysfunction with ageing. Indeed, degradation and reduced activity of HSFs in ageing and role of HSP90, HSP70 in life span extension reveal the functional status of chaperones in ageing process [52].
As discussed earlier, misfolded proteins are preliminary aspect for poly (Q) disorders and molecular chaperones have been implicated as key regulators of protein aggregation [53]. It was found that expression and inducibility of HSPs in ageing brain is drastically reduced. In addition, evidences like similarity in reduction of HSPs level, HSPs inducing transcription factors, HSF-1 in human ageing brain and various animal disease models further highlight the probable mechanistic link between ageing and poly (Q) pathology [54,55]. Progressive decline of HSP70 and HSP40 co-chaperones HDJ1 and HDJ 2 and several synaptic chaperones have been detected in aged HD animal models [56]. However, significant role of HSPs in HD pathology is unclear and controversial.
The probable roles of chaperones in disease suppression have been widely investigated in various animal models. Not surprisingly, overexpression of HSPs ameliorates the disease toxicity and reduced age related cellular impairment. Overexpression of HSP70, HJD1 in Drosophila MJD and HD model demonstrate the capability of HSPs in disease suppression by restoring axonal transport, cell death and ultimately, life span extension [49]. Investigation in poly (Q) models of S. cerevisiae, C. elegans and mouse further validate the importance of HSP70 and HSP40 in regulating poly (Q) aggregation and toxicity [57,58]. In order to find out the potential mechanism for progressive reduction of HSPs in poly (Q) disease, several mechanisms have been hypothesized, including transcriptional deficit of HSPs expression via the toxic poly (Q) protein and sequestration of cellular soluble HSPs along with the toxic aggregates to form IBs. Evidences like transcriptional impairment of HSP70 in Drosophila model, role of CBP via reduction of HSF-1 activity, further support the transcriptional deficit hypothesis [59].
In addition to the chaperones, importance of ubiquitinproteosome system (UPP) and lysosome-mediated autophagy in protein homeostasis and their role in disease onset is a matter of concern. Decrease in UPP system and autophagic-lysosomal system with ageing have been reported and correlated with disease onset [60]. Although correlation between reduced autophagy and poly (Q) aggregation in HD model has been demonstrated but exact role of UPP and autophagic-lysosomal system remain elusive [59,61]. It appears that mis-regulation of these several factors which are responsible for protein quality control might be the risk factor for disease occurrence and therefore, proper maintenance of cellular protein homeostasis will provide a new strategy to delay disease onset and ageing.
Several years ago, discovery of daf-2 (worm homolog of insulin receptor) and age-1 (catalytic subunit of PI3K) in C. elegans laid the foundation that ageing is a genetically regulated process and manipulation in longevity pathway can potentially delay ageing related changes. Mutations in daf-2 and age-1 extend life span more than double times in worms [62]. Subsequent research lead to the discovery of Insulin/insulin-like growth factor signalling pathway (IIS) and its role in regulating daf-2, age-1 and daf-16 in longevity.
Insulin/insulin-like growth factor signalling pathway is evolutionary conserved across the phyla in worms, flies, mice and humans and is the best characterized ageing signalling network [63]. Insulin/insulin-like growth factor receptor, DAF-2/IR mediates its downstream cascade via the activation of kinases like PI3K and AKT [64], which negatively regulate transcription factors, DAF-16/FOXO, SKN-1 and HSF-1 through phosphorylation [63,65]. Initiation of insulin/IGF-1 signalling cascade results in nuclear exclusion of FOXO/ DAF-16, SKN-1 and HSF-1 interacting partner DDL-1. FOXO/DAF- 16, SKN-1 and HSF-1 being transcription factors regulate subsets of target genes that play important roles in ageing and longevity of the organisms. In the absence of insulin/IGF-1 cascade, downstream transcription factors including FOXO/DAF-16, HSF-1 are translocated into the nucleus, where they activate expression of their downstream target genes including chaperones. Loss of daf-2 and age-1 slows down several cellular ageing related changes, through modulation of longevity pathways [66]. Expression analysis by using DNA microarrays, Serial Analysis of Gene Expression (SAGE), Chromatin immunoprecipitation (ChIP) in mutant vs wild type has revealed overlapping target genes between daf-16 and HSF. Similar to FOXO/DAF-16 and HSF-1, SKN-1 was also found to regulate longevity by counteracting oxidative stress.
Above finding in C. elegans has led to the investigation of similar genes in other organisms including humans. Mutation in dInR (Drosophila insulin like receptor) extends life span through similar regulation of dFOXO (Drosophila homologs of human FOXO) [67]. Moreover, over expression of dfoxo in fat body and mutation of chico (Drosophila insulin receptor substrate) extends life span and increased stress resistance in flies [68,69]. Similar functional consequences have been reported in mammalian systems as reduced insulin/IGF- 1 signalling cascade extends life span and delay the ageing processes. Knockout model of IGF-1R (IGF-1 receptor), FIRKO (adipose tissue specific insulin receptor) in mice system, have demonstrated consequent increase in life span [70]. Study from klotho (kl) gene, has found to repress insulin/IGF-1 signalling cascade and further provide the evidences of insulin pathway in regulating ageing, as over expression of kl results in life span extension [71]. Conservation of insulin/IGF-1 signalling pathway in regulating ageing process from invertebrate to mouse model further raised its potential role in regulation of human ageing. Not surprisingly, recent studies have suggested that genetic variants of human klotho gene and insulin signalling molecules are associated with ageing and extended life span. Further mutation in FOXO3A (human homolog of DAF-16) are found to increase longevity [62]. IGF-1 receptor mutation has been identified in centenarians from different ethnic groups and thus, correlating the increase in longevity and reduction of IGF-1 signalling [72].
Focusing on the disease associated stress condition, it was interesting to investigate the role of insulin/IGF-1 signalling in poly (Q) aggregation and toxicity as ageing is the key factor for disease onset. The direct link of insulin/IGF-1 signalling in poly (Q) aggregation has come from C. elegans studies when it was demonstrated that insulin/ IGF-1 also neutralizes the poly (Q) aggregation and toxicity in HSF-1 and DAF-16 mediated manner and protect the worms from motility impairment caused by poly (Q) toxicity [73]. Machado-Joseph disease (MJD) model of poly (Q) disease also revealed disaggregation of toxic aggregates by reducing insulin/IGF-1 signalling pathway [74]. Above studies clearly indicate that insulin/IGF-1 signalling pathway plays a crucial role in poly (Q) toxicity suppression through the modulation of ageing process by reducing proteotoxicity in disease. The remarkable property of insulin/IGF-1 signalling in neuroprotection has also been demonstrated in several studies of mouse HD and AD models. Reduction of IGF-1 receptor and IRS (Insulin Receptor Substrate) in mouse knockout models rescues the animal from behavioural impairments, learning and memory deficit [64]. Collectively, these mammalian studies indicate neuroprotective function of insulin/IGF- 1 signalling via modulation of ageing processes and key attention for therapeutic development in future.
Dietary restriction and nutrient-sensing signalling cascade
Dietary Restriction (DR) is the phenomenon of life span extension by reduced nutrient intake without malnutrition. It appears that the role of DR in regulation of ageing and longevity is conserved across the phyla including yeast, invertebrates and mammals [75]. Effect of DR on longevity creates a centre of attention for researchers to dissect out the underlying biological mechanisms. Decades of studies have explored various nutrient sensing pathways as the key player in DR induced life extension, such as Sirtuin and TOR (Target of Rapamycin) pathways, under the indirect control of insulin/IGF-1 signalling cascade.
Sirtuin (Sir2), NAD-dependent deacetylase protein is a key mediator in DR induced life span extension that is found to be conserved in yeast, worms, flies and mammals [76]. For the first time, Sir2 has been discovered from yeast system as an important molecule necessary for DR induced longevity. Subsequently, effect of Sir2 in life span has been demonstrated in worms, flies and mice models as extra copies of Sir2 extend 50% life span in worms, 30% in Drosophila and in mice, Sirt6 knockout accelerate ageing [77-79]; thus highlighting the conserved role of Sir2 in mediating the beneficial effect of DR on animal life span and behaviour. Human genome poses seven homolog of yeast Sir2, Sirt1-7. Subsequently, occurrence of polymorphic variants of Sirt in human centenarians suggests conservation of Sirtuin function from yeast to human. Interestingly, yeast deletion mutation of Sir2 aggravates poly (Q) aggregation and toxicity giving a clue of DR response in neutralizing disease pathology through delayed ageing [80]. Similarly, in C. elegans, Sirtuin pathway has been linked to poly (Q) toxicity and ageing as extra copies of Sir2 in worm increases longevity and also suppresses poly (Q) mediated toxicity. Chemical compound such as Resveratrol, extends life span and neutralized poly (Q) mediated toxicity in Sir2 dependent manner in invertebrates and mammalian model by mimicking DR [81]. Moreover, Resveratrol, an activator of Sir2 has been demonstrated to increase life span in C. elegans and Drosophila [65,81]. Taken together, understanding the underlying mechanisms of DR in poly (Q) toxicity and ageing modulation are emerging as an exciting areas for future exploration to develop novel therapeutic strategies.
Similar to Sirtuin signalling cascade, TOR pathway also has potential role in DR response and studies in yeast, worms, flies and rodent models demonstrated its potential role in longevity and conservation across the phyla. Reduced activity of TOR in yeast system activates the transcription factor Gis1, which mediate the expression of superoxide dismutase (Mn-SOD), an antioxidant that acts as a scavenger for cellular superoxide free radicals [82]. Moreover, increased free radicals in ageing TOR mutant yeast further establish its role in clearance of free radicals. In general, TOR pathway regulates translation through activation of S6 kinase (S6K) and inhibition of translational repressor eIF4EBP. Knockdown of the homolog of three translational regulators eIF4EBP and S6K in C. elegans extends worm life span but the exact mechanism is still not clear [70]. Similarly, deregulation of translation in Drosophila either by TOR dominant mutant or mutation of its downstream target S6K extends life span [83]. Interestingly, it was also demonstrated that shortening of life span in Drosophila due to dFOXO mutation has been compensated by DR which highlights the intersection of Insulin/IGF-1 pathway and TOR pathway in controlling animal ageing process [84]. Despite having much of this information, the precise roles of TOR in aging and poly (Q) disorders are not well understood. Since various stress conditions like oxidative stress plays central role in disease pathology and ageing, it would be interesting to further investigate the role of TOR in animal longevity and disease pathogenesis.
Mitochondrial dysfunction and redox-homeostasis in ageing
Mitochondrion, being a central organelle in regulation of energy homeostasis controls some of the crucial cellular process like energy metabolism, production and presence of Reactive Oxygen Species (ROS) and cellular apoptosis which are important in determining life span and disease pathogenesis [85]. It has been hypothesized that defects in mytochondrial dynamics due to accumulation of mutation in mitochondrial DNA (mtDNA) may contribute to abrupt increase in ROS levels. This sudden increase in cellular ROS may cause damage to biologiacal macromolecules mainly DNA and protein, consequently affecting cellular protein homeostasis which is generaly associated with ageing and neurodegenarative disorders [59,74]. The brain tissue, because of its high energy demand, become more vulnerable to the increased ROS due to inefficient mitochondrial function [86]. Likewise, several recent evidences suggest an intimate correlation between mitochondrial dysregulation and increase in ROS level in enhanced ageing and disease susceptibility. Mutations affectng respiratory Electron Transport System (ETC) system or growth in low oxygen environment increase longevity in C. elegans by decreasing cellular ROS level [59]. In addition, RNAi mediated knockdown of several components of ETC in C. elegans, including NADH/ ubiquinone redox oxidoreductage, cytochrome-c reductase and cytochrome-c oxidase as well as mitochondrial ATP synthase extend life span [74]. Similar effect of ETC in life span extention via the ROS dynamics has also been reported in mouse model. However, further investigation is required in order to explore the intimate role of mitochondrial respitatory system in regulation of ageing and longevity.
Rise in oxidative stress is usually associated with the poly (Q) mediated disease onset [87]. It has been proposed that overload in antioxidant mechanisms might be responsible for increasing oxidative damage and cellular ROS level in poly (Q) expanded disease [56,57]. Elevated level of oxidatively damaged biomacromolecules including damaged DNA, protein and lipid in poly (Q) mouse model and HD pateints indicate ROS as one of the key players in intervening the molecular protein homeostasis during disease pathogenesis.
Despite having much of these information and identification of basic molecular and cellular pathways that play important role in disease pathogenecity, the precise mechanisms of neurotoxicity caused by poly (Q) expanded protein is not well understood. Figure 2 provides a collective network of various factors affecting ageing and neurodegeneration and also attempts to establish a possible link between the two. Studies on the fundamental developmental networks clearly indicate involvement of common signalling pathways during ageing and neurotoxicity, and therefore, it will be interesting to explore the mechanistic link between ageing and poly (Q) mediated neurotoxicity in future to understand the disease pathogenecity in greater detail and to develop novel therapeutic stratigies.
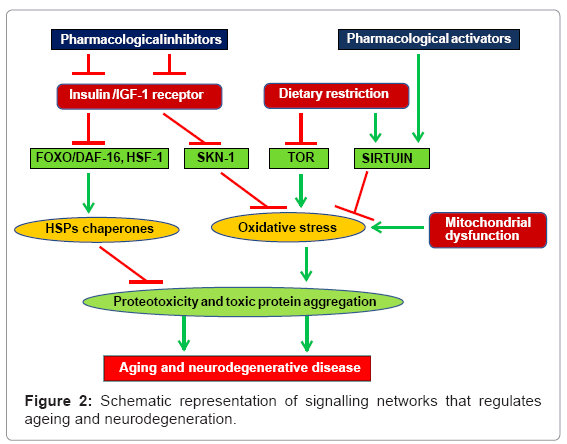
Figure 2: Schematic representation of signalling networks that regulates ageing and neurodegeneration.
Therapeutic approaches to ameliorate poly (Q) disorders
Extensive research on poly (Q) and other neurodegenerative diseases proposed several therapeutic strategies to cure these fatal illnesses. Most of such approaches attempt to decrease pathogenicity either by targeting mutant proteins and protein aggregates or reduces the toxic effects caused by these aggregates. Strategies to ameliorate poly (Q) disorders include enhancing mutant protein degradation, inhibition of protein aggregate formation, modifying altered transcription, channelizing mitochondrial energy metabolism and antioxidants usage, preventing excitotoxicity and use of antiapoptotic drugs [88-90]. None of above however, is targeting the mechanism involved or the root cause of the diseases and till date there is no promising medical therapy available for mitigating the clinical manifestations of poly (Q) diseases. This part of the review is focused on some upcoming strategies which act upstream in disease pathogenesis and could be further refined to achieve the goal of defeating extremely daunting neurodegenerative disorders.
Cell therapy aims to replenish the neuronal cells those are degenerated during the course of disease progression [91]. Extensive work has been performed on transplanting foetal-derived cells into the brain of HD patients. Several clinical trials performed on mice and humans diseased brain suggest that transplanted foetal striatal allografts can well survive and establish functional connections to host tissues, but this can only delay the disease progression and does not drive a complete rescue [92-96]. Stem cells transplantation appears to have more therapeutic potential than fetal-derived cells because of the minimal chances of host rejection. In addition, stem cells therapy also has advantage of triggering endogenous repair mechanism and protecting host neuronal cells through trophic support [97,98]. Neural Stem Cells (NSC) could be derived from fetal cells or adult brain cells. It can also be proliferated by culture technique. After transplantation, NSCs have been found to adjust well in host brain circuitry by developing morphological and electrical properties similar to mature neurons [99]. In numerous models of neurodegeneration, grafted NSC exhibited the property of migration towards the site of neuronal degeneration where they differentiate and rebuilt the lost synaptic connections [98,100]. In lesion induced impairment tests, NSC treatment has shown partial to full recovery [98,101].
Although the most significant property of NSC is to form neuronal cells in host brain but their propensity to differentiate into non neuronal cells such as astrocytes cannot be overlooked. It has been proposed that these non-neuronal cells may increase the efficacy of stem cell therapy in HD by performing neuroprotective role in degenerative environment, as it is believed that in HD, the key factors driving neurodegeneration are the aberrations in the neurons’ surrounding microenvironment rather than the inherent neuronal properties [91]. Subsequently, several reports indicate that astrocytes might interact with NSC to promote their propagation and differentiation thus helping newly formed neuron to integrate in brain neural circuit to make functional mature neuron [102].
Simultaneously, attempts were also made to use NSC for providing deficit growth factors to protect degenerating neurons. These factors cannot be given to patients in form of drugs as they cannot cross bloodbrain barrier being larger in size. Hence, utilizing NSC to provide these growth factors to degenerating neurons as a support is being examined. Transplanted stem cells can serve as, a source of various Neuronal Growth Factors (NGF), such as Brain Derived Neutrotrophic Factor (BDNF), Glial Cell Derived Neurotrophic Factor (GDNF) and Ciliary Neurotrophic Factor (CNTF) [103]. Transgenic mouse model has shown considerable improvement in motor functions when its striatal and hippocampal BDNF scarcity was supplemented [104]. Such results are motivating as it demonstrates that enhanced level of BDNF via cell therapy can significantly improve the motor function in HD models [91].
Stem cell therapy is emerging as a promising tool since risk of tumour formation posed by stem cells could be lessened by treating the cells with growth factors in vitro before grafting them into host [105]. However, it has to be realized that cell therapy is in infancy and a detail understanding of stem cell biology need to be developed in order to improve the efficacy of this potential remedial method.
Invention of induced Pluripotent Stem Cells (iPSCs) is one of the pioneering prospects of cell therapy that may resolve the issue of limited access to availability of stem cells. It was demonstrated that somatic cells can be reprogrammed to form pluripotent stem cells (iPSCs) by inducing the expression of four or even less transcription factors [106]. It was observed that if fibroblasts or any differentiated human somatic cells transduced with four viral vectors each carrying the gene for transcription factors Oct4, Sox2, Klf-4 and c-Myc, respectively then these cells can be induced to differentiate into stem cells [107]. With a further development, different cell types both of mouse and humans like hepatocytes, dermatocytes have been successfully reprogrammed to differentiate into iPSCs [108,109]. However, a lot of studies need to be performed to address few safety issues like tendency of iPSCs to form tumours, use of viral delivery vectors etc., before final clinical applications for therapeutic purposes.
As discussed earlier, as exact mechanism of poly (Q) toxicity is still not well understood so targeting upstream events i.e. before formation of nuclear IBs by therapeutic strategies is quite useful. One such methodology that has been extensively worked by several groups involve silencing of mutated mRNA using RNA based strategies such as Antisense Oligonucleotides (AONs) and RNA interference (RNAi).
The RNA therapy exploits the inherent mammalian RNA based gene silencing pathway where primary microRNA (miRNA) hairpin structures are transcribed by specialized non coding regions. Further, miRNA is chopped to short double stranded RNAs by a chain of enzyme complexes and then antisense strand of these duplex RNAs is loaded on RNA-induced silencing complex (RISC) [110]. RISC mediates mRNA degradation or translation suppression of complementary target mRNA, depending upon accuracy of base pairing [111]. This kind of therapy could be executed using either short interfering RNA (siRNAs) or short hairpin RNAs (shRNAs). siRNAs are synthetic double stranded RNA molecules of approximate length of 21 nucleotides having sequence complementary to mRNA targeted for silencing [112]. shRNAs pose slight complexity in its structure as the nucleotides are positioned into a stem loop region and are transcribed under strong polymerase III promoter ensuring its constitutive expression. This may exhaust the cellular RNAi machinery leading to disruption of endogenous miRNA pathway resulting in vivo toxicity [113]. It could be circumvented by designing modified shRNAs having endogenous features of primary miRNAs for its regulated expression [114].
AON approach provides an alternative approach for silencing target mRNA. AONs are synthetic short antisense 15-25 long oligonucleotides that bind to target in a sequence specific manner and silence the mRNAs either via RNAse H facilitated degradation or by blocking translation through steric hindrance posed to translational machinery [115-117].
The following strategies can be undertaken for utilizing RNA therapy in neurodegenerative disorders:
i. Non-allele-specific gene silencing
Therapeutic studies undertaken during last few years attempted using a technically simple non-allele-specific gene silencing methodology [118]. This strategy involve introduction of an exogenous mutated copy of gene in organisms already having a normal copy [119]. To check the efficiency of effector molecules such as siRNAs and AON in repressing the mutated transgene was examined rather than inspecting their capability to differentiate between exogenous mutated and endogenous wild alleles [120].
In mouse model of SCA1, neurodegeneration was reduced following delivery of RNAi effectors against human mutant Atx1 transgene [121]. As reported earlier, absence of wild-type Htt has lethal effects but it was found that targeted silencing of Htt in different HD models could ameliorate the disease phenotype [120]. In HD-N171-82(Q) mice model, mutant human transgene and endogenous wild type Htt levels were reduced by 75% by using miRNA based gene silencing which resulted in improvement of motor coordination in mice. Although, silencing of Htt results in transcriptional suppression of a large number of endogenous genes. Similar transcriptional deregulations in transgenic HD models further support the belief that HD toxicity is contributed by loss-of-function of proteins entrapped in IBs [119]. All these reports and many others seem to prove that silencing of both, mutant and wild type mRNA could be tolerated without severe signs of toxicity.
In future, it will be essential to discern the threshold levels of wild type protein required by cell for circumventing the deadly effects posed due to the loss of wild type protein. Moreover, in many poly (Q) disorders, lack of wild type protein proved to be fatal at many times.This strengthens the need of allele-specific silencing approach as wild type proteins are necessary for cell viability in many cases.
ii. Allele-specific gene silencing
Allele-specific gene silencing requires designing of silencing effector molecules those can differentiate between wild and mutant allele of disease causing gene. It aims to achieve maximum target selectivity of mutant transcript by effector molecule and simultaneously avoiding wild type transcript from being silenced. To avoid silencing of wild type allele, at least one nucleotide mismatch should be incorporated in effector molecules.
In poly (Q) disorders, wild type and mutant allele differs in their respective number of CAG repeats. Particularly, this is challenging for small 22-25 nucleotides long effector molecules to distinguish between the two. Moreover, abundance of CAG repeats throughout the genome enhances the complexity [122]. In this respect, presence of SNP linked to CAG repeats in a particular group of patients could be exploited for implementation of RNAi based allele-specific gene silencing [112]. So, identification of SNPs linked to the diseased gene is most crucial stage while designing the effector molecule. However, many neurodegenerative disorders have been reported to harbour easily distinguishable nucleotide mismatch between wild and mutant allele [123,124].
Following screening of desired SNP, RNAi effector molecules could be designed to optimize them for targeting mutant allele-specific transcript. It is suggested that purine:purine mismatches in the target sequence results strong disruption of siRNA-mRNA duplex [125]. It has also been observed that mismatches in 3` region of siRNA specially between 10-16 nucleotides confers more strong binding to target mRNA [126].
Allele-specific gene silencing depending on single nucleotide difference has been shown to be operative in many neurodegenerative disorders [123,127,128]. This approach has been attempted in three poly (Q) disorders; directed to SNPs in case of Atxn3 and Atxn7, and in HD, it targets deletion in mutant Htt [122]. Interestingly, it was demonstrated that compare to 40% degradation of mutant transcripts, only 7% of wild type transcripts were degraded after targeting triplet deletion present in mutant Htt allele [129].
This is an alternative approach to allele-specific gene silencing where wild type protein function is retained. In this strategy, initially both the wild type and mutant gene transcripts are silenced using regular non-allele-specific gene silencing and to retain the wild type protein expression, host is injected with codon optimized copy of wild type gene which is resistant to silencing [125]. The extra copy of gene is made resistant to silencing by modifying its sequence by deleting siRNA recognition sites and trying to retain all the amino acid coding sequences [130].
In a cell based model of SCA6, function of wild-type protein was restored by transfecting cells with siRNA targeting both wild and mutant transcript of Cacna1A, and an extra copy of wild type gene provide resistant to siRNA [131]. A similar type of study was also performed in mouse model of amyotrophic lateral sclerosis (ALS) to regain the function of wild type Sod1 protein [132]. Gene targeting is another strategy to replace the mutant copy of a disease gene with its wild counterpart.
In gene targeting, mutated diseased allele is exchanged to wild type allele in-situ using the cellular mechanism of Homologous Recombination (HR). In this technique, a short stretch of wild type allele of mutated gene provided in vector is exchanged on the basis of sequence homology [133]. The main obstacle in implementation of this therapy is highly incompetent efficiency of HR i.e. (∼1 in 106 cells) [134]. Probably this is because of an alternative pathway- non homologous end joining (NHEJ) which is more favoured in somatic cells causing random integration of desired transgene [135]. Nevertheless, stem cells favour HR over NHEJ increasing the relevance of exploiting stem cells in gene targeting [136].
Majority of work that has been performed in this area has been either performed on mouse or human stem cells (HSCs and ESCs, respectively). In this strategy, human stem cells are extracted from diseased individual and amended in-vitro for the disease causing mutation. Lastly, ameliorated healthy stem cells are transplanted back into the affected body part of the host. This approach utilizes autologous cells which nullifies the possibility of transplant rejection in the host. This therapy has been successfully implemented in curing sickle cell anaemia in mouse model [137]. Although gene targeting has not been tested in any of the poly (Q) models, however, since these disorders are mostly monogenic, there are splendid possibilities that this strategy could be potentially useful in management of neurodegenerative disorders.
Although above stated approaches appear technically convincing, there are number of issues in the way of implementing these therapies. Acquisition and availability of HSCs is a difficult deed and ESCs faces ethical issues. Also, ESCs can be rejected by host being non autologous. Moreover, HR frequency for mutation repair is still very low in stem cells, raising the need to use viral vectors for transgene transfer which upsurges the safety issues in genome integrity. Fortunately, because of advancement in research, new avenues are unleashing like introduction of zinc finger nucleases (ZFNs) which may potentially facilitate the use gene targeting successfully in curing neurodegenerative disorders.
Zinc finger nucleases are genetically engineered fusion proteins hrhaving two functional domains– zinc finger domain and endonuclease domain [134]. Zinc finger domains are generally found in transcription factors and are responsible for binding of ZFNs to the major grove of DNA to make contact with the nucleotide triplets. Another, endonuclease domain of ZFNs confers it with the ability to cut phosphodiester bond on DNA backbone to make a Double Strand Break (DSB) [138]. Hence, ZFNs are tailor made proteins that can create DSB at specific DNA sequence. As gene targeting was limited by low frequency of HR, recently a group has achieved a HR frequency of up to 20% for targeted gene replacement in a disease causing gene in human cell lines using ZFNs [139]. It has been demonstrated that HD mouse model injected with zinc finger drug in brain could bind to targeted mutated DNA sequence and can halt the polymerase from transcription of mutated diseased RNA. There was 50% reduction in the gene activity at the site of drug injection and the protein inclusions were also reduced by 40% in diseased mouse [140]. This approach is beneficial over gene silencing because this does not target diseased mRNA; rather transcription of diseased mRNA is arrested.
While it is increasingly clear now that neurodegeneration and ageing is delimited by several explicit yet common signalling pathways/ factors, how these various pathways/ factors control life span and influence ageing or triggers/modulate neurodegeneration is still enigmatic. The theatrical progress made in recent years utilizing novel strategies and various model organisms has demonstrated the feasibility of decoding this enigmatic relation between ageing and neurodegeneration and further studies are expected to provide exciting insights.
Research programmes in laboratory is supported by grants from the Department of Biotechnology (DBT), Government of India, New Delhi; DU/DSTPURSE scheme and Delhi University R & D fund to SS.