Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2015) Volume 4, Issue 3
The human clinical trials with seleno-methionine (SeMet) for prostate cancer prevention and selenized-yeast (contains mostly SeMet) for the prevention of non-small cell lung cancer and prostate cancer in North America conclusively rejected the use of these selenium (Se) forms for cancer prevention in human populations with adequate Se intake. Nevertheless, solid mechanism-based preclinical studies with other Se forms have suggested the potential for their use at pharmacological doses as adjuvant treatment alone and especially as chemo-enhancers for combination cancer therapy. Of the distinct pools of Se metabolites, the mono-methylated Se (MM-Se) has many desirable attributes for cancer therapy, affecting a multitude of crucial molecules and signaling pathways in cancer epithelial cells, vascular endothelial cells and microenvironment. Inorganic selenite/selenide in excess of selenoprotein synthesis can lead to DNA single strand breaks, which implicate possible genotoxicity to normal cells and are therefore unattractive for longterm use. In this paper, we review animal studies with MM-Se such as methylseleninic acid and Se-methylselenocysteine as well as inorganic Se for inhibition of xenograft tumors of several organ sites as single therapy and those using MMSe to enhance the therapeutic efficacy of cancer chemotherapeutic drugs and to reduce the dose-limiting toxicities of such modalities. We present and critique potential mechanisms of action for such applications and future improvement. Since Se-methylselenocysteine was the only MM-Se with a published human pharmacokinetic study, we discuss future research directions to enable clinical translation studies.
<AR: Androgen Receptor; ER: Estrogen Receptor; Se: Selenium; MM-Se: Mono-Methyl(Ated)-Selenium; DMDSe: Di-Methylselenide; GPx3: Glutathione Peroxidase 3; Ph-SeGPx: Phospholipid Glutathione Peroxidase; MSeA: Methylseleninic Acid; MSeC: Se-Methylselenocysteine; MSeCN: Methylselenocyanate; SeCys: Selenocysteine; Se-GPx: Glutathione Peroxidase; Sel-P or SEPP1: Selenoprotein P; Sel-W: Selenoprotein W; SeMe: Selenomethionine; TDI: Thyroixine Deiodinase; Trx: Thioredoxin; TrxR: Thioredoxin Reductase; GR: Glutathionine Reductase
By now, several well-designed human clinical trials in North America have conclusively dismissed using seleno-methionine (SeMet) or SeMet-rich selenized-yeast for cancer prevention in human populations with adequate Se intake [1-4]. These failures to show preventive efficacy of SeMet and Se-yeast have cast shadow on the potential utility of other and newer forms of Se (e.g., monomethylated Se, MM-Se) for cancer chemoprevention. Many reviews have been published discussing the reasons for the failure [5-8]. Based on the well-documented metabolic and biochemical differences between SeMet and the MM-Se [9,10], as well as the preclinical efficacy outcomes [11], we have argued that the failure of SeMet cannot be equated to all Se forms are ineffective for cancer chemoprevention [12]. We have recently articulated two major lessons from these trials [13]: 1) Antioxidant hypothesis was tested in wrong subjects/patient populations; 2) Se agent selection was not supported by cell culture and animal efficacy data.
Whereas the major focal point of Se research for the last two decades has been on the cancer preventive efficacy of SeMet and Se-Yeast in human trials, several groups, including those of Y. Rustum and C. Ip at Roswell Park Cancer Institute and ours, have studied the possible cancer therapeutic application, especially of the MM-Se forms distinct from SeMet. In this paper, we review animal studies with MM-Se such as methylseleninic acid (MSeA) and Se-methylselenocysteine (MSeC) as a single agent in “adjuvant therapy” context and those using MM-Se to enhance therapeutic efficacy of chemotherapy drugs in a number of cancers and to reduce dose-limiting toxicities of such drugs. We examine and contrast pertinent studies with inorganic Se forms such as selenite and selenite in the context of cancer therapy. In addition, we present and critique potential mechanisms of action for therapeutic applications and for their further refinements. Furthermore, we discuss the current status of human studies with MM-Se and suggest future research directions to enable clinical translation studies. Because we focus on endogenous Se metabolism and MM-Se metabolites, we will not discuss Se-substituted derivatives of S-containing drugs or aromatic synthetic Se compounds. Readers interested in these topics are referred to following expert reviews [8,14].
Active Anti-Cancer Metabolite Theory
Se deficiency is not a health concern in the USA. As a result, most animal models and cell culture studies since the mid-1980s have dealt with chemotherapeutic or chemopreventive levels of Se and have focused on the cancerous cells as the targets of its anti-cancer effects. Most animal models have shown cancer chemopreventive activity of Se intake that is 20-50 times greater than the nutritional requirement [10]. It had been proposed decades ago that cancer chemoprevention by Se in nutritionally adequate subjects was independent of the antioxidant activity of plasma or tissue glutathione peroxidase (SeGPx) [10,15]. This paradigm was based on the observation that the dietary level of Se (2 ppm or greater as selenite or other Se forms) needed to achieve a significant cancer preventive activity in rodent animal models far exceeded that required (i.e., 0.1 ppm or mg/kg) to support maximal SeGPx in the blood (glutathione peroxidase 3, GPx3) or the target tissues from which the experimental cancers arose. This view has been extended to the other selenoproteins identified subsequently in the past 2 decades, including phospholipid glutathione peroxidase (Ph-SeGPx or GPx4), selenoprotein P (Sel-P or SEPP1), selenoprotein W (Sel-W), thyroixine de-iodinases (TDI) and thioredoxin reductases (TrxRs) [9,16,17]. The studies with transgenic suppression of selenoproteins (increased prostate and colon cancer risk with decreased SeGPxs in the presence of adequate dietary selenite) and the TrxR1 knockdown transfectant cells (decreased lung cancer growth with knocked down TrxR1) indicate likely contradicting roles of these proteins as regulators of cancer risk in the nutritional range of Se intake [18].
Metabolically, excess Se beyond the need for selenoprotein synthesis (hydrogen selenide is co-translationally incorporated into the selenocysteine (SeCys)-containing selenoproteins) is methylated into methylselenol, which is further methylated and excreted as dimethylselenide (volatile through breath) and trimethylselenonium (urine) or converted into selenosugars (Figure 1). Although SeMet is the most abundant natural MM-Se, it is predominantly incorporated into general proteins in place of methionine (non-specific substitution) or metabolized to SeCys through a trans-selenation pathway similar to the transulfuration pathway from methionine to cysteine. The efficiency of the latter pathway will be dependent on the metabolic capacity of the cell types and organs. Liver and hepatocytes are expected to be well equipped with the metabolic enzymes, whereas in general non-hepatic tumor cells in culture would be expected to be limited in this ability.
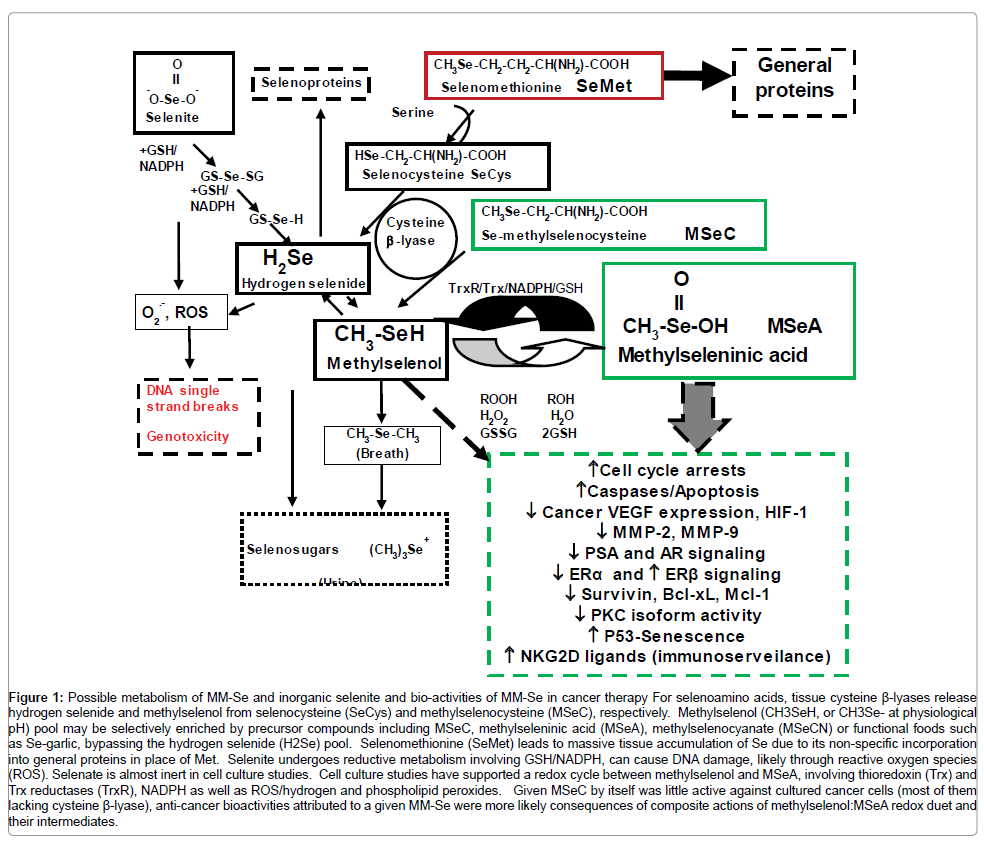
Figure 1: Possible metabolism of MM-Se and inorganic selenite and bio-activities of MM-Se in cancer therapy For selenoamino acids, tissue cysteine β-lyases release hydrogen selenide and methylselenol from selenocysteine (SeCys) and methylselenocysteine (MSeC), respectively. Methylselenol (CH3SeH, or CH3Se- at physiological pH) pool may be selectively enriched by precursor compounds including MSeC, methylseleninic acid (MSeA), methylselenocyanate (MSeCN) or functional foods such as Se-garlic, bypassing the hydrogen selenide (H2Se) pool. Selenomethionine (SeMet) leads to massive tissue accumulation of Se due to its non-specific incorporation into general proteins in place of Met. Selenite undergoes reductive metabolism involving GSH/NADPH, can cause DNA damage, likely through reactive oxygen species (ROS). Selenate is almost inert in cell culture studies. Cell culture studies have supported a redox cycle between methylselenol and MSeA, involving thioredoxin (Trx) and Trx reductases (TrxR), NADPH as well as ROS/hydrogen and phospholipid peroxides. Given MSeC by itself was little active against cultured cancer cells (most of them lacking cysteine β-lyase), anti-cancer bioactivities attributed to a given MM-Se were more likely consequences of composite actions of methylselenol:MSeA redox duet and their intermediates.
Ip and Ganther [10,19] proposed that the active anti-cancer Se metabolites were likely MM-Se species (presumably methylselenol) and the cancer chemopreventive efficacy of a given Se compound might depend on the rate of its metabolic conversion to the active MM-Se form(s). Circumstantial evidence was obtained by comparing the efficacy of forms of Se that fed into different Se metabolite pools, with precursors of methylselenol displaying greater preventive efficacy than those for hydrogen selenide or dimethylselenide in the chemically induced rodent mammary carcinogenesis model [20,21]. Extending on the methylselenol structure-activity theme, subsequent work had shown that the alkyl-selenol and allyl-selenol precursor compounds were more active against mammary carcinogenesis than methylselenol precursors on an equal molar basis of dietary Se intake [22,23]. However, these structure-activity studies have not been extended beyond the mammary carcinogenesis model for assessing the general applicability of the methylselenol hypothesis in other organ sites.
In cell culture models, studies by us and others focusing on the levels of Se exposure in therapeutic range have shown that MM-Se compounds that are putative precursors to the methylselenol pool induce numerous cellular, biochemical and gene expression responses that are distinct from those induced by the forms of Se that enter the hydrogen selenide pool [9,16]. These major cellular and biochemical effects have been summarized and detailed in earlier reviews [9,16]. We updated newer findings from the last decade and schematically present them in Figure 1.
Hydrogen selenide pool
Sodium selenite and sodium selenide, which feed into the hydrogen selenide (H2Se) pool, rapidly (within minutes to a few hours of Se exposure) induce DNA single strand breaks (SSBs), S phase or G2/M cell cycle arrest and lead to subsequent cell death by apoptosis, autophagy and necrosis [9]. Sodium selenide and SeCys could recapitulate the DNA SSB induction and the apoptosis effects of selenite in the model system [24]. A superoxide dismutase mimetic compound, copper dipropylsalicylate, blocked DNA SSBs and apoptosis, indicating that selenite per se did not trigger these events [25]. Studies have provided further support for ROS (superoxide generation) as intermediates for activating p53 Ser-15 phosphorylation in apoptosis induced by selenite in LNCaP prostate cancer cell model [26,27]. Published data from our group indicated that selenite given by daily oral dosage of 3 mg/kg body weight to tumor bearing nude mice increased DNA SSBs in peripheral blood nucleated cells whereas, the same dosage of MSeA or MSeC lacked this effect [11]. Further studies in animal models and in humans are necessary to confirm the in vivo genotoxicity of selenite.
The methylselenol pool
Our group has shown that MM-Se methylselenol precursors such as methylselenocyanate (MSeCN) and MSeC induced apoptosis of mammary tumor epithelial cells and leukemia cells without the induction of DNA single strand breaks (SSBs) [24,28,29]. Furthermore, MSeA-induced cancer cell apoptosis was caspase-dependent, whereas selenite-induced cell death was independent of these death proteases in DU145 prostate cancer cells with mutant P53. MM-Se led to G1 arrest [28-33], inhibitory effects on cyclin-dependent kinases [33,34] and protein kinase C [35]. In terms of genotoxicity implications, a daily oral dosage of 3 mg per kg body weight, MSeA and MSeC significantly suppressed human DU145 xenograft growth without increasing DNA SSBs in the peripheral blood nucleated cells of the host mice whereas the same dosage of selenite caused increased DNA SSBs and was ineffective for suppressing xenograft growth [11].
Direct demonstration of methylselenol intra-cellular cytotoxicity through adenoviral delivery of bacterial Pseudomonas putida methioninase (Ad-METase) gene into cancer cells was carried out by R. Hoffman’s group in vitro and in vivo, when SeMet was provided as substrate to enzymatically produce methylselenol [36]. In METasetransduced tumor cells, the cytotoxicity of SeMet is increased by 3 orders of magnitude compared with non-infected cells. A strong bystander effect occurred because of methylselenol release from METaseproducing cells and uptake by surrounding tumor cells. They showed that methylselenol damaged the mitochondria via oxidative stress and caused cytochrome c release into the cytosol, thereby activating the caspase cascade and apoptosis. In animal xenograft model, they showed that Ad-METase/SeMet treatment profoundly inhibited tumor growth in mice and significantly prolonged their survival. The same group [37] also investigated the combination of SeMet/Ad-METase gene therapy with doxorubicin in a nude mice model with H460 lung cancer cell xenograft. Doxorubicin was administered by intraperitoneal injection (i.p.) twice per week at a dose of 2 mg/kg body weight and SeMet of 1 μmole (i.e., 79 μg) per mouse by intratumoral injection daily, starting the day after the adenovirus infection. After two weeks of treatment, the control- and the doxorubicin-treated tumors had increased by ten times. The tumors in mice treated by SeMet/Ad-METase grew 5.8 times, and those in mice receiving a combination of doxorubicin and SeMet/Ad-METase grew only 2.5 times. It was also determined that the tumor doubling time of the respective individual treatment was around 2-3 days but was increased to 10 days by the combination treatment.
Our group demonstrated that methylselenol generated from SeMet via METase added directly to cell culture medium efficiently induced cell apoptosis [32] in human umbilical vein endothelial cells (HUVECs) and the DU145 human prostate cancer cells. Exposure of DU145 cells to methylselenol generated in the sub-micromolar range of substrate SeMet led to caspase-mediated cleavage of poly(ADPribose) polymerase (cPARP), nucleosomal DNA fragmentation, and morphologic apoptosis, recapitulating MSeA effects cited above. Biochemically, METase/SeMet-generated methylselenol also inhibited phosphorylation of protein kinase AKT and extracellularly regulated kinases 1/2 (ERK 1/2) as did MSeA. In the HUVECs, methylselenol exposure resulted in G1 arrest action similar to MSeA in mitogen stimulated G1 progression during mid-G1 to late G1. This stage-specificity was also mimicked by inhibitors of phosphatidylinositol 3-kinase. In the last 5 years, Zeng and coworkers in a number of publications [38-40] followed up this means of generating methylselenol in other cancer cell culture models and examined cell cycle and molecular changes. Overall, these findings indicate that methylselenol is one of the proximal active metabolites.
Beside apoptosis, we and others have shown that MM-Se compounds exert a rapid inhibitory effect on the expression of key molecules of cancer and angiogenesis. For example, sub-apoptotic concentrations of MSeA inhibited the expression and secretion of the angiogenic factor VEGF in several cancer cell lines [41]. MSeA also inhibited the expression of matrix metalloproteinase (MMP)- 2 in the vascular endothelial cells [41,42]. These effects plus a potent inhibitory effect on the cell cycle progression of vascular endothelial cells [31,32] indicate that methylselenol can be a key inhibitor of angiogenic switch regulation in early lesions and in tumors [9]. MSeA and methylselenol released by METase from SeMet inhibited androgen receptor (AR) expression and its signaling to prostate specific antigen (PSA) [43-45] as well as PSA stability [43]. MSeA has also been shown to inhibit estrogen receptor (ER) signaling in breast and endometrial cancer cells [46-49] and as a novel suppressor of aromatase expression [50]. Specifically, the expression of the aromatase gene, CYP19, is controlled in a tissue-specific manner by the alternate use of different promoters. In obese postmenopausal women, increased peripheral aromatase is primarily attributed to the activity of the glucocorticoid stimulated promoter, PI.4, and the cAMP-stimulated promoter, PII. MSeA effectively suppressed aromatase activation by dexamethasone (a synthetic glucocorticoid), and forskolin (a specific activator of adenylate cyclase). Unlike the action of aromatase inhibitors, MSeA suppression of aromatase activation is not mediated via direct inhibition of aromatase enzymatic activity. Rather, it is attributable to a marked down-regulation of PI.4- and PII-specific aromatase mRNA expression, and thereby a reduction of aromatase protein.
In primary human fibroblast cell culture MSeA was shown to induce cellular senescence, an irreversible arrest of cell proliferation, more potently than other Se forms by W. Cheng’s group [51,52]. They found that MSeA, MSeC and selenite at concentrations less than or equal to their respective LD50 induced senescence only in the noncancerous cells MRC-5 (lung fibroblasts) and CRL1790 (colon fibroblasts), not in cancer cell lines. They showed that this senescence induction by MSeA was dependent on ATM kinase activity and wild type p53 in the MRC-5 cells, as inhibition or genetic silencing of ATM or p53 decreased induction of senescence.
In addition to apoptosis and senescence induction, MSeA was shown by our group to induce autophagy in some but not all pancreatic cancer cell lines [53]. Effort to attenuate autophagy signaling led to increased apoptosis in MSeA-treated cells, consistent with a generalized notion that drug-induced autophagy confers survival advantage to the cancer cells. Our finding stresses the multiple-targeting nature of MSeA exposure and the cellular fate will be determined by the balance of these, oftentimes opposing, signaling pathways.
Extending on the MM-Se specificity theme, MSeA, MSeC and dimethylselenide (DMDSe) were observed to stimulate the cell surface expression of ligands for the lymphocyte receptor NKG2D in Jurkat T cells [54], specifically inducing the expression of MICA/B major histocompatibility complex class I related chain genes, which are upregulated in stressed cells for immune system recognition. MSeA and DMDSe up-regulated the maximum MICA/B response at 5 μM, On the other hand, selenite, selenate, SeMet, selenocysteine and hydrogen selenide had no effect on the cell surface expression of MICA/B. At the transcription level, MSeA and MSeC induced the mRNA levels of MICA/B and ULBP2, whereas selenite did not. The work suggests MMSe could improve NKG2D-based cancer immune therapy.
Methylselenol ←→MSeA redox cycle through thioreduxin reductase (TrxR)
Gromer and Gross [55] examined Ganther’s speculation [19] that methylselenol and MSeA might exert their effects by inhibition of the selenoenzyme TrxR via the irreversible formation of a diselenide bridge. Failing to find such Se entity, they showed that MSeA did not act as an inhibitor of mammalian TrxR but was an excellent substrate, which was reduced by TrxR according to the equation 2 NADPH + 2 H+ + CH3SeO2H → 2 NADP+ + 2 H2O + CH3SeH. They identified the Se-containing product of this reaction by mass spectrometry using silver to trap the reactive methylselenol. Nascent methylselenol was found to efficiently reduce both H2O2 and glutathione disulfide GSSG. They found that MSeA was a poor substrate for human glutathione reductase (GR, not a selenoprotein) and the catalytic selenocysteine residue of mammalian TrxR was essential for MSeA reduction to methylselenol.
R. Gopalakrishna’s group showed [56] that MSeA, but not methylselenol, inactivated protein kinase C (PKC) isoforms which depending on the type, site and extent of modification could be involved in either tumor progression or promotion. MSeA was shown to inactivate pure PKC enzyme activity, which could be reversed by the TrxR system or thiol agents, but methylselenol did not. In two prostate cancer cell lines (DU145 and LNCaP) under serum-starved conditions, MSeA (5 μM ) caused the reduction in PKC activity as early as 5 to 15 minutes. PKC activity recovered slightly by 2 hours but not to the level of the control activity. The extent of inactivation in these cell lines were observed to be less than in that of the pure PKC enzyme possibly due to TrxR-mediated MSeA←→MSeH redox cycling to remove MSeA from the site of action. The increase in PKC inactivation, in particular the promitogenic and prosurvival PKCepsilon isoenzyme was observed to be associated with increased apoptosis and cell growth inhibition. For other protein kinase enzymes, for example, protein kinase A, 10 times higher MSeA was required to inactivate. The inactivation of PKC was further enhanced when MSeA was made from methylselenol by the PKC-bound phospholipid peroxides within close proximity to PKC thioclusters. As low as nanomolar Se resulted in the oxidation of the catalytic unit of PKC by the MSeA-methylselenol redox cycle, highlighting the specificity of MSeA in inactivating PKC.
In a subsequent paper by the same group [57], MSeA was shown in submicromolar concentrations to prevent the transformation of prostate epithelial cells. However, micromolar levels of MSeA were required to prevent cell growth, invasion and cause apoptosis in prostate cancer cells. MSeA sensitivity was inversely correlated to PKCepsilon levels, with PKCepsilon ectopic over-expression resulting in minimum MSeA induction of epithelial cell transformation and prostate cancer cell apoptosis. In addition, resistance to MSeA treatment was linked to increased TrxR expression and inhibition of TrxR increased the cancer cells’ sensitivity to MSeA. These studies suggest that both PKCepsilon and TrxR can negate the efficacy of MSeA.
Therefore, it could be that methylselenol, MSeA and their redox cycling intermediates, especially in localized protein microenvironment of thioclusters, may provide specific targeting niches to negatively regulate enzymatic activities involved in cancer promotion or growth. Retrospectively, many of the reported activities that we and others attributed to the “methylselenol” pool should be recast in the framework of dynamic MSeA:MSeH redox cycling balances (Figure 1).
In vivo “molecular targets” of MM- Se in cancer models
Our group examined the impact of acute Se treatments (i.e., daily single oral gavage of 2 mg Se /kg of body weight for 3 days) of female Sprague-Dawley rats bearing 1-methyl-1-nitrosourea-induced mammary carcinomas to increase the probability of detecting in vivo apoptosis and the associated gene/protein changes in the malignant epithelial cells [58]. Whereas control carcinomas doubled in volume in 3 days, MSeC and selenite treatments caused regression in approximately half of the carcinomas, accompanied by a 3- to 4-fold increase of apoptosis and approximately 40% inhibition cancerous epithelial cell proliferation. The mRNA levels of growth arrest-DNA damage inducible 34 (gadd34), gadd45, and gadd153 genes were, contrary to expectation [29], not higher in the Se-treated carcinomas than in the gavage- or diet restriction-control groups. The Gadd34 and Gadd153 proteins were localized in the non-epithelial cells and not induced in the cancer epithelial cells of the Se-treated carcinomas. On the other hand, both Se forms decreased the expression of cyclin D1 and increased levels of p27Kip1 and c-Jun NH2-terminal kinase activation in a majority of the mammary carcinomas. In addition, the lack of induction of gadd genes in vivo by MSeA was confirmed in a human prostate xenograft model in athymic nude mice. In summary, these experiments showed the induction of cancer epithelial cell apoptosis and inhibition of cell proliferation by Se in vivo through the potential involvement of cyclin D1, p27Kip1, and c-Jun NH2-terminal kinase pathways. They cast doubt on the three gadd genes as mediators of Se action in vivo.
Using the transgenic adenocarcinoma mouse prostate (TRAMP) model, our group established the efficacy of MSeA and MSeC against prostate carcinogenesis and characterized potential mechanisms [12]. Eight-week-old male TRAMP mice (C57B/6 background) were given a daily oral dose of water, MSeA, or MSeC at 3 mg Se/kg body weight and were euthanized at either 18 or 26 weeks of age. By 18 weeks of age, the genitourinary tract and dorsolateral prostate weights for the MSeAand MSeC-treated groups were lower than for the control (P < 0.01). At 26 weeks, 4 of 10 control mice had genitourinary weight > 2 g, and only 1 of 10 in each of the Se groups did. In addition, Se treatment resulted in delayed lesion progression, increased apoptosis, and decreased proliferation without appreciable changes of T-antigen expression in the dorsolateral prostate of Se-treated mice. Decreased serum insulinlike growth factor I when compared with control mice was observed in the Se-treatment groups as well. In another experiment, giving MSeA to TRAMP mice from 10 or 16 weeks of age increased their survival to 50 weeks of age, and delayed the death due to synaptophysinpositive neuroendocrine carcinomas and synaptophysin-negative prostate lesions and seminal vesicle hypertrophy. Wild-type mice receiving MSeA from 10 weeks did not exhibit decreased body weight or genitourinary weight or increased serum alanine aminotransferase compared with the control mice. Therefore, these Se compounds were effective in the inhibition of this model of prostate carcinogenesis.
Our proteomic analyses suggest unique potential molecular targets for each of these chemoprevention-active MM-Se forms with little “protein targets” overlap between MSeA and MSeC [59] and they are not interchangeable. Additionally, we detected a possible adverse prostate cancer risk profile for MSeC through onco-proteins such as fatty acid synthase. These data, when considered in the framework of dynamic MSeA:MSeH redox cycling, would be reasonable pending on the entry point of MM-Se to fuel the redox cycle. Whether such findings are present in mammary and other organ sites should be examined to assess the generalizability. Further investigations of the tissues/organs exposed to these two MM-Se forms in higher mammal species such as dogs and primates may help to predict and inform the potential adverse impacts of each form for human translation.
MM-Se forms are superior to SeMet or selenite in a therapy context
Whereas both MSeC and MSeA have been shown to inhibit mammary carcinogenesis in the 1990’s (21, 60), their anti-cancer efficacy in prostate or other non-mammary organs has only recently been tested. In several in vivo models, the MM-Se compounds in comparison with SeMet or selenite were more efficacious in reducing tumor sizes and reducing molecular markers. Our group has shown that the orally administered MSeA and MSeC dose-dependently (1 and 3 mg Se/kg) inhibit the in vivo growth of DU145 human prostate cancer xenograft in athymic nude mice whereas selenite and SeMet are not active [11]. Each Se was given by a daily single oral dose regimen starting the day after the subcutaneous inoculation of cancer cells (resembling residual and disseminated cancer cells in an adjuvant chemotherapy context). In the same study, MSeA was observed to be more active than MSeC against PC-3 xenograft growth. In terms of tolerability, all four Se compounds at the tested doses of 3 mg per kg or lower did not adversely affect the body weight of the mice. Selenite treatment did however increase DNA single-strand breaks in peripheral lymphocytes, whereas the other Se forms did not. The measurement of Se content in the tissue showed that SeMet treatment led to 9.1-fold more liver Se retention and approximately 3.6 times higher than mice treated with an equal dose of methyl-Se, even though this form of Se was least effective in reducing the DU145 or PC-3 xenograft growth. The observed massive tissue Se accumulation (non-specific incorporation in place of Met into proteins) and the lack of anti-cancer potency agreed well with earlier work with SeMet in conventional rodent models [21,61]. In summary, MSeA exhibited superior in vivo “adjuvant” therapy efficacy against two human prostate cancer xenograft models over SeMet and selenite, without the genotoxic property of selenite.
Yan and Demars have shown that MSeA at 2.5 mg Se/kg provided in AIN93G diet significantly reduced pulmonary metastatic yield when compared with the controls (p<0.05), SeMet did not have such an effect [62] using Lewis lung carcinoma (LLC) in male C57BL/6 mice. Mice were fed AIN93G control diet or that diet supplemented with MSeA or SeMet at 2.5 mg Se/kg for 4 weeks at which time they were injected intramuscularly or subcutaneously with 2.5 × 105 LLC cells. Experiments were terminated 2 weeks later for mice injected intramuscularly or 2 weeks after surgical removal of primary tumors from mice subcutaneously injected with cancer cells. Dietary supplementation with MSeA significantly reduced pulmonary metastatic yield when compared with the controls (p < 0.05) in both models; however, SeMet did not. MSeA significantly decreased plasma concentrations of urokinase-type plasminogen activator (p<0.05) and plasminogen activator inhibitor-1 (p<0.05) and vascular endothelial growth factor (p<0.05), fibroblast growth factor (p<0.05) and platelet derived growth factor-BB (p < 0.05), SeMet did not affect any of the aforementioned measurements. These results demonstrate that MSeA reduces spontaneous metastasis of LLC in mice, perhaps through inhibition of the urokinase plasminogen activator system and reducing angiogenesis.
Combination therapy applications of MM-Se vs. SeMet in animal models
A few studies in the last decade have resulted in a renewed interest in the therapeutic potential of Se as an enhancer of existing treatment modalities. Y. Rustum’s group [63] used athymic nude mice bearing human non-small cell carcinoma HNSCC (FaDu and A253) and colon carcinoma (HCT-8 and HT-29) xenografts to evaluate the potential role of Se compounds as selective modulators of the toxicity and antitumor activity of selected anticancer drugs: fluorouracil, oxaliplatin, cisplatin, taxol and doxorubicin with particular emphasis on irinotecan, a topoisomerase I poison. They showed that a sub-lethal dose of Se either as MSeC or SeMet was highly protective against toxicity induced by these chemotherapeutic agents. Furthermore, MSeC significantly increased the cure rate (no detectable tumor at the transplant site for up to 3 months after treatment was terminated) of xenografts bearing human tumors that are sensitive (HCT-8 and FaDu) and resistant (HT-29 and A253) to irinotecan. An increased cure rate (100%) was achieved in nude mice bearing HCT-8 (20% with irinotecan alone) and FaDu xenografts (30% with irinotecan alone) treated with the MTD of irinotecan (100 mg/kg/week for 4 weeks) when combined with MSeC. Administration of higher doses of irinotecan (200 and 300 mg/kg/week for 4 weeks) was required to achieve high cure rate for HT-29 and A253 xenografts. Administration of these higher drug doses was possible due to selective protection of normal tissues by Se. The observed in vivo protective action against drug toxicity was highly dependent on the schedule of Se, which required a minimum of 3 days ahead of the first drug treatment.
In their next study [64], the effect of MSeC on the pharmacokinetic and pharmacogenetic profiles of genes relevant to irinotecan metabolic pathway to identify possible mechanisms associated with the observed combinational synergy was evaluated. Nude mice bearing tumors (FaDu and A253) were treated with MSeC, irinotecan, and their combination. Samples were collected and analyzed for plasma and intra-tumor concentration of irinotecan and its active form 7-ethyl-10-hydroxylcamptothecin (SN-38) by high-performance liquid chromatography. After MSeC treatment, the intra-tumor concentration of SN-38 increased to a significantly higher level in A253 than in FaDu tumors and was associated with increased expression of carboxyesterase CES1 (involved in the de-esterification of irinotecan) in both tumor models. MSeC/irinotecan treatment, compared with irinotecan alone, resulted in a significant decrease in levels of ABCC1 and DRG1 (multi-drug resistant associated proteins, drug efflux pumps) in FaDu tumors and an increase in levels of CYP3A5 and TNFSF6 (involved in increased drug metabolism and inducing apoptosis respectively) in A253 tumors. No statistically significant changes induced by MSeC/irinotecan were observed in the levels of other investigated variables (transporters, degradation enzymes, DNA repair, and cell survival/death genes).
In a subsequent paper [65], the Rustum group further examined the basis for MSeC in increasing the therapeutic index of irinotecan against human tumor xenografts using the FaDu and A253 models. A MSeC minimum effective dose of 0.01 mg/day for 28 days to the maximum tolerated dose (MTD) of 0.2 mg/day for 28 days was established for enhancing efficacy, with treatment beginning 7 days prior to irinotecan treatment. From the lower MseC doses to the MTD, the mice’s cure rate in the combination with irinotecan was increased. On its own, MSeC did not have any effect on the cure rate but reduced tumor growth by as much as 30%. The highest plasma Se concentration was achieved 1 hour after a single dose and 28 days after daily treatment of MSeC. The ability of FaDu tumors to retain Se was significantly better than A253 tumors, and the highest Se concentration in normal murine tissue was achieved in the liver. Peak plasma and tissue Se concentrations were functions of the dose and duration of MSeC treatment. The MSeC-dependent increase in Se level in normal murine tissues may contribute to the protective effect against irinotecan toxicity observed in those tissues. Intra-tumoral total Se concentration was not found to be predictive of the combination therapy response rates. These authors pointed out a critical need to develop a method to measure the active metabolites of MSeC, rather than total Se.
The same group [66] demonstrated that one mechanism of selectivity was the differential impact of MSeC on the content of irinotecan and its active metabolite SN-38 between tumors of HNSCC and the normal tissue. In this situation, the in vivo synergy between MSeC and irinotecan is influenced by treatment schedule. For the FaDu tumors, the concurrent combination (MSeC and irinotecan administered for 2 hours) resulted in no increase in the enhancement of irinotecan’s response rate. However, the sequential combination of MSeC administered for 7 days before irinotecan resulted in 65% increase in irinotecan’s response rate. These findings were also seen in the A253 xenografts. The combination of MSeC/irinotecan enhanced tumor vessel maturation, intra-tumor concentration of SN-38 and apoptotic death of tumor cells. Normal tissue drug concentrations were not impacted by MSeC treatment. Their finding is of clinical relevance for using MSeC to decrease tumor drug resistance and achieve higher active metabolite of irinotecan to ultimately enhance cure rates.
Our group established the enhancement of paclitaxel efficacy by MSeA in androgen receptor-negative PCa [67]. In nude mice, the paclitaxel and MSeA combination inhibited growth of the DU145 subcutaneous xenograft with the equivalent efficacy of a four-time higher dose of paclitaxel alone. MSeA decreased the basal and paclitaxelinduced expression of Bcl-XL and survivin in vitro and in vivo. Ectopic expression of Bcl-XL or surviving attenuated MSeA/paclitaxel-induced apoptosis. The sensitization effect of MSeA on paclitaxel has been confirmed in a triple-negative breast cancer xenograft model [68]. The synergism was attributable to more pronounced induction of caspase-mediated apoptosis, arrest of cell cycle progression at the G2/M checkpoint, and inhibition of cell proliferation. Treatment of SCID mice bearing MDA-MB-231 triple-negative breast cancer xenografts for four weeks with MSeA (4.5 mg/kg/day, orally) and paclitaxel (10 mg/kg/week, through (i.p.) resulted in a more pronounced inhibition of tumor growth compared with either agent alone. The combination of MSeC with estrogen receptor positive breast cancer chemotherapy drug tamoxifen also resulted in synergistic tumor growth inhibition in the MCF-7 breast xenograft tumors in ovariectomized female athymic nude mice [69]. Sustained-release estradiol was implanted into the ovariectomized mice, which allowed the MCF-7 tumors to grow. Tamoxifen pellets were also implanted subcutaneously while MSeC was administered by i.p. after the tumors reached 100 mm3. For the tumors in mice given estradiol implant, tamoxifen and MSeC had the greatest suppression, while each agent on its own had some suppression. At termination of the study, cell proliferation and angiogenesis were reduced as early as seven days by MSeC.
Y. Dong’s group aimed to exhibit the efficacy of MSeA and a recently approved androgen receptor antagonist drug MDV3100 (Enzalutamide) both in vitro and in vivo as well as the MSeC and MDV3100 combination in vivo [70]. Using prostate cancer 22Rv1 cells in androgen-deprived conditions, dihydrotestosterone (DHT)- stimulated trans-activating activity of the androgen receptor was suppressed by the combination of MSeA and MDV3100 in the most statistically significant manner as compared to each individual agent. In addition, the mRNA levels of the androgen receptor downstream targets PSA and KLK2 had the most pronounced inhibition both with and without DHT with the MDV3100 and MSeA combination. The effect of these two compounds in inhibiting cell growth was found to be synergistic with the combination treatments producing combination indexes of less than 1. In the 22Rv1 tumor xenograft model, however, the results were quite different. The MDV3100 dose used was 10 mg/ kg and 3 mg Se/kg/day for both MSeC and MSeA. The tumor growth of the animals being treated with both MSeA and MDV3100 showed no difference from the group treated only with MDV3100. On the other hand, the group treated with the combination of MSeC and MDV3100 had the smallest tumors, with them being significantly smaller than any of the single agent treatments. The authors offered the explanation that the lack of MSeA and MDV3100 efficacy in vivo might be due to MSeA and MDV3100 conjugation when prepared in the same dosing solution.
Cisplatin (CDDP) use in oncology is largely limited by its severe side effects including gastrointestinal toxicity and nephrotoxicity. For testing the utility of sodium selenosulfate to attenuate CDDP side effect, Li and co-workers treated mice by i.p. with 9 μmol sodium selenosulfate/ kg for 11 days [71]. On days 5 and 7, they gave the mice an injection of CDDP of 8 mg/kg 1 hour after sodium selenosulfate treatment. Sodium selenosulfate decreased the incidence of diarrhea as a measure of gastrointestinal toxicity from 88% to 6%. Such a prominent protective effect promoted them to evaluate the safety potential of long-term sodium selenosulfate application in comparison with sodium selenite. Mice were administered with each Se for 55 days at the doses of 12.7 and 19 μmole/kg (1.0 mg and 1.5 mg Se/kg by i.p. injection). The lowdose sodium selenite caused growth suppression and hepatotoxicity which were aggravated by the high-dose, leading to 40% mortality rate, but no toxic symptoms were observed in the two sodium selenosulfate groups. Their results suggest sodium selenosulfate at an innocuous dose can markedly prevent CDDP-induced gastrointestinal toxicity while improving cancer “cure” rate.
Potential mechanisms of efficacy enhancement
Using androgen-independent and p53 non-functional prostate cancer cell culture models, our group investigated the Se specificity and signaling pathways underlying the enhancement action on apoptosisinduced by different classes of chemotherapeutic drugs [72]. DU145 and PC3 human AR-negative PCa cells were exposed to minimal apoptotic doses of Se and/or the topoisomerase I inhibitor SN38 (irinotecan active metabolite), the topoisomerase II inhibitor, etoposide or the microtubule inhibitor paclitaxel/taxol. The results showed that sublethal MSeA increased the apoptosis potency of SN38, etoposide, or paclitaxel by several folds higher than the expected sum of the apoptosis induced by MSeA and each drug alone. The combination treatment did not further enhance JNK1/2 phosphorylation that was induced by each drug in DU145 cells. The JNK inhibitor SP600125 substantially decreased the activation of caspases and apoptosis induced by MSeA combination with SN38 or etoposide and completely blocked these events induced by MSeA/paclitaxel. A caspase-8 inhibitor completely abolished apoptosis and caspase-9 and caspase-3 cleavage, whereas a caspase-9 inhibitor significantly decreased caspase-3 cleavage and apoptosis but had no effect on caspase-8 cleavage. None of these caspase inhibitors abolished JNK1/2 phosphorylation. In contrast to MSeA, selenite did not show any enhancing effect on the apoptosis induced by these drugs. The results support the enhancing effect was primarily through interactions between MSeA and JNK-dependent targets to amplify the caspase-8-initiated activation cascades in a p53- defective background.
In a follow up study, our group established the enhancement of paclitaxel efficacy by MSeA in vivo, and investigated Bcl-XL and survivin as molecular targets of MSeA to augment apoptosis in PCa [67]. MSeA decreased the basal and paclitaxel-induced expression of Bcl-XL and survivin in vitro and in vivo. Ectopic expression of Bcl-XL or survivin attenuated MSeA/paclitaxel-induced apoptosis. Along the line of suppression of survival molecules, MSeA was shown to enhance ABT-737 apoptosis in several cancer cell lines: breast, prostate and colon cancer cell lines [73]. Potential mechanisms were attributed to the decreased Mcl-1 (prosurvival molecule) expression by MSeA both at the basal level and due to ABT-737 induction, in addition to the reactivation of Bad, a pro-apoptotic protein after its inactivation by ABT- 737 by MSeA. The synergistic effect was dependent on Bax expression in the model system, suggesting a central role of mitochondria apoptosis.
Rustum’s group examined synergistic activity in the clonal TRAMP cell line C2G by MSeC and docetaxel [74]. Cells were treated with combinations of MSeC and/or docetaxel concurrently or sequentially with 24 hours MSeC pretreatment. It was observed that the concurrent administration of MSeC and docetaxel in this cell line did not enhance docetaxel’s efficacy. On the other hand, the 24 hour pretreatment of MSeC enhanced docetaxel’s cell growth inhibition synergistically. Using this treatment scheme, caspase-3 activity was significantly increased as early as 30 minutes. The caspase inhibitor z-VAD-fmk significantly attenuated the increased apoptosis induced by the combination treatment, indicating that the synergistic apoptosis is caspase-dependent. Survivin was decreased significantly by the combination treatment as compared to each drug individually.
In several breast cancer cell lines, MSeA with tamoxifen synergistically increased the caspase-mediated apoptosis as observed by the increased cleavage of caspases -7, -8 and -9 and PARP [75]. Cytochrome c and Bim were also increased due to MSeA treatment. The inhibition of the caspases by the general caspase inhibitor completely blocked MSeA induced apoptosis on its own and with tamoxifen. Specific caspase -8 and -9 inhibition suggested that the cleavage of caspase 9 was needed for the cleavage of caspase-8 by MSeA.
In cell culture models, the MSeA-specific enhancement action on drug-induced apoptosis was also found with tumor necrosis factorrelated apoptosis-inducing ligand (TRAIL). Yamaguchi et al. [76] demonstrated that the concomitant treatment with TRAIL and MSeA produced synergistic effects on the induction of apoptosis in androgendependent LNCaP and androgen-independent DU145 prostate cancer cells. MSeA rapidly down-regulated the expression of the cellular FLICE inhibitory protein, a negative regulator of death receptor signaling. In addition, they demonstrated that the synergistic effects of MSeA and TRAIL resulted from the activation of the mitochondrial pathwaymediated amplification loop. MSeA also effectively blocked TRAILmediated BAD phosphorylation at Ser112 and Ser136 in DU145 cells and was accompanied by induction of the mitochondrial permeability transition and the release of cytochrome c and Smac/DIABLO proteins from the mitochondria and into the cytosol. These results suggest that MSeA may help to enhance efficacy of and overcome resistance to drug-induced or TRAIL-mediated apoptosis in prostate cancer cells.
Whereas p53 was not required for the enhancement effect of MSeA on apoptosis induced by drugs or TRAIL as discussed above [72,76], our group has shown a critical role of p53 and Bax/mitochondria pathway of caspases to mediate selenite’s ability to enhance apoptosis induced by TRAIL in the LNCaP cells [27]. Selenite induced a rapid generation of superoxide and p53 Ser-15 phosphorylation, an indicator of DNA damage. It also increased Bax abundance and translocation into the mitochondria. Selenite and TRAIL combined treatment led to synergistic increases of Bax abundance and translocation into mitochondria, loss of mitochondrial membrane potential, cytochrome c release and the cleavage activation of caspases-9 and -3. Inactivating p53 with a dominant negative mutant abolished apoptosis without affecting superoxide generation, whereas a superoxide dismutase mimetic agent blocked p53 activation, Bax translocation to mitochondria, cytochrome c release and apoptosis induced by selenite/TRAIL. In support of Bax as a crucial target for crosstalk between selenite and TRAIL pathways, introduction of Bax into p53-mutant DU145 cells enabled selenite to sensitize these cells for TRAIL-induced apoptosis. The results indicate that selenite induces a rapid superoxide burst and p53 activation, leading to Bax up-regulation and translocation into mitochondria, which restores the crosstalk with stalled TRAIL signaling for a synergistic caspase-9/3 cascade-mediated apoptosis execution.
It is therefore possible that the p53 functional status of the cancer may influence the choice of Se forms to provide the most enhancement of efficacy to be balanced with an optimal reduction of side effects. Since the risk for selenite-induced DNA damage and genotoxicity in the treatment of a cancer patient is less of a concern than for primary prevention use, the combined use of selenite and methyl Se with chemotherapeutic drugs may target a broader spectrum of cancers.
Rustum’s group investigated the role of MSeC on increased drug delivery via tumor vascular maturation in mice with FaDu head and neck squamous cell carcinoma (HNSCC) xenografts after 2 weeks of oral MSeC treatment [77]. Changes in microvessel density (CD31), vascular maturation (CD31/alpha-smooth muscle actin), perfusion (Hoechst 33342/DiOC7), and permeability (dynamic contrast-enhanced magnetic resonance imaging) were determined at the end of the 14-day treatment period. Double immunostaining of tumor sections revealed a marked reduction ( approximately 40%) in microvessel density accompanying tumor growth inhibition following MSC treatment along with a concomitant increase in the vascular maturation index ( approximately 30% > control) indicative of increased pericyte coverage of microvessels. Hoechst 33342/DiOC7 staining showed improved vessel functionality, and dynamic contrastenhanced magnetic resonance imaging using the intravascular contrast agent, albumin-GdDTPA, revealed a significant reduction in vascular permeability following MSC treatment. They found a 4-fold increase in intratumoral doxorubicin levels with MSC pretreatment compared with administration of doxorubicin alone. Similar conclusion was reached with a different drug irinotecan [66] in that its efficacy was influenced by treatment schedule with MSeC and associated with enhancement of tumor vessel maturation, intra-tumor concentration of active metabolite SN-38 and apoptotic death of tumor cells. Normal tissue drug concentrations were not impacted by MSeC.
This group further examined the mechanism of enhanced irinotecan efficacy by MSeC in HNSCC with respect to the angiogenic master regulator hypoxia inducible factor 1 (HIF-1) [78]. MSeAinduced down regulation of or shRNA knockdown of HIF-1α, which is the upstream-regulator of VEGF and carbonic anhydrase IX (CAIX), resulted in the increased cell death under hypoxic but not normoxic conditions. In the animal model, the combination treatment of irinotecan and MSeC in treating the parental xenografts in comparison with the HIF-1α knockdown tumors treated with only irinotecan resulted in similar therapeutic efficacy, supporting a role of HIF-1a as a target of MSeA to enhance therapeutic effect.
In a separate study from the Rustum group [79], the phase II drug conjugating enzyme Ugt1a, which metabolizes lipophilic molecules into water soluble metabolites, was observed to be necessary for MSeC’s protective efficacy against toxicity caused by irinotecan in rats. In the Ugt1a mutant rats, the maximum tolerated doses of irinotecan were lower than wild-type rats. This was specific for irinotecan as no differences were observed for docetaxel and cisplatin which are not substrates for Ugt1a.
In summary, MM-Se compounds (likely through methylselenol: MSeA redox cycle) enhance therapeutic efficacy through several potential mechanisms of action, ranging from increased tumor vascular maturity and drug delivery and retention, reduction in the expression of prosurvival molecules, increased caspase-mediated apoptosis, to improved tolerance to toxicity of the chemotherapeutic drugs, which usually have dose-limiting toxicity (Figure 2). These results indicate that there is a possible role for MM-Se compounds in enhancing the therapeutic efficacy of the current approved drugs and their efficacy, scheduling optimization and mechanisms of action should be further investigated.
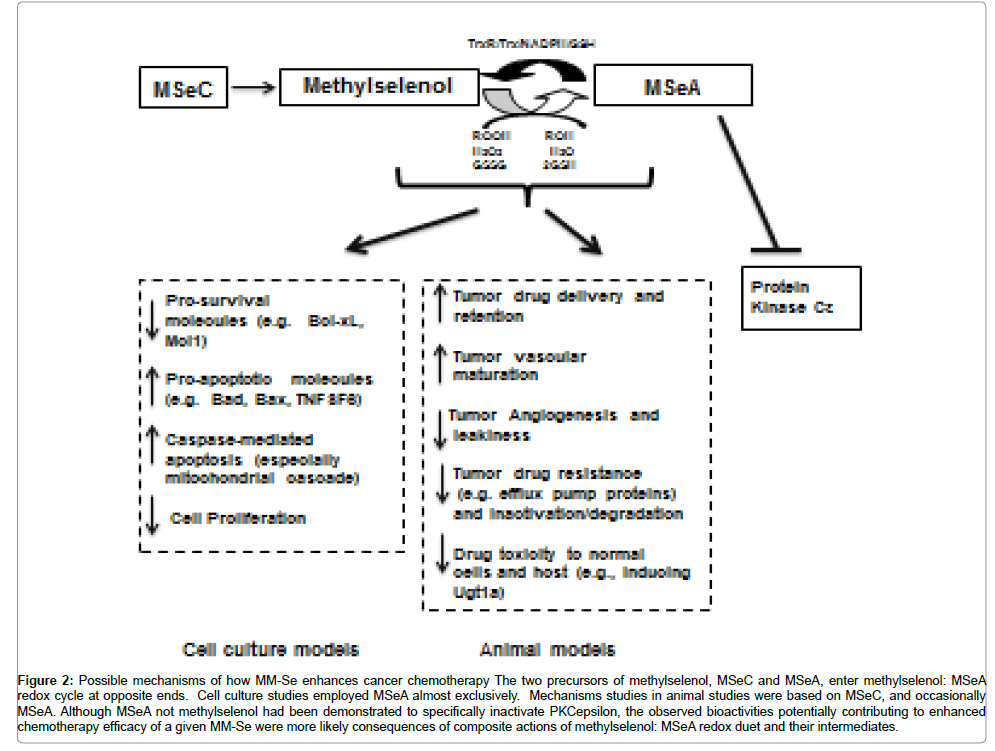
Figure 2: Possible mechanisms of how MM-Se enhances cancer chemotherapy The two precursors of methylselenol, MSeC and MSeA, enter methylselenol: MSeA redox cycle at opposite ends. Cell culture studies employed MSeA almost exclusively. Mechanisms studies in animal studies were based on MSeC, and occasionally MSeA. Although MSeA not methylselenol had been demonstrated to specifically inactivate PKCepsilon, the observed bioactivities potentially contributing to enhanced chemotherapy efficacy of a given MM-Se were more likely consequences of composite actions of methylselenol: MSeA redox duet and their intermediates.
Translational studies involving Se for cancer therapy
The observed improvement of MTD for a number of chemotherapeutic drugs in rodent models by MSeC and SeMet led Rustum and co-workers [80] to conduct a phase I study to determine the impact of a fixed, non-toxic high dose of SeMet on the MTD of irinotecan in cancer patients. SeMet was given orally as a single daily dose containing 2.2 mg of Se starting 1 week before the first dose of irinotecan. The Se dosage was 11 times higher than that used in the Clark study [81] or SELECT. Irinotecan was given by i.v. once weekly every 6 weeks (one cycle) for 4 cycles. The starting dose of irinotecan was 125 mg/m2/week. Escalation (by 30% each time) occurred in cohorts of three patients until the MTD was defined. Pharmacokinetic studies were done for Se, irinotecan and irinotecan’s metabolites. The results showed that three of four evaluable patients at dose level 2 of irinotecan (160 mg/m2/week) had dose-limiting diarrhea. None of the six evaluable patients at dose level 1 (125 mg/m2/week irinotecan) had dose-limiting toxicity. One patient with a history of irinotecan-refractory colon cancer achieved a partial response. SeMet displayed a long half-life of prolonged accumulation towards steady-state concentrations. SeMet did not significantly change the pharmacokinetics of irinotecan or any of its metabolites. However, the co-administration of SeMet significantly reduced the irinotecan biliary index, which has been associated with gastrointestinal toxicity. It was also shown that the plasma concentrations of Se were sub-optimal. The authors concluded that SeMet at the dose and schedule used did not allow for the safe escalation of irinotecan beyond the previously defined MTD of 125 mg/m2. Disease stabilizations were noted in this highly refractory population. Given the outcomes of SELECT and HGPIN (Progression From High-Grade Prostatic Intraepithelial Neoplasia to Cancer) studies (3, 4), the lack of effects is not surprising in hindsight. Considering the better action profiles of MM-Se than SeMet discussed above, it will be very interesting to consider MSeC and MSeA for future trials.
This group also performed a study to determine the recommended dose of SeMet amongst seven tested doses in combination with irinotecan to achieve selenium concentrations of over 15 μM after 1 week of SeMet loading [80]. A total of 31 patients were enrolled and evaluated for treatment-related toxicity. They all had a confirmed solid tumor for which therapy was not available for treatment. SeMet was administered twice a day orally during the loading phase in the form of 400 or 800 μg capsules with dosages from 3200 to 7200 μg. Irinotecan was adminstered after a week of SeMet treatment at a fixed dose of 125 mg/m2 once weekly. In spite of the Se concentrations surpassed 15 μM, SeMet did not offer any protection against irinotcan toxicity.
An Australian group assessed the safety, tolerability and pharmacokinetics of sodium selenate in men with castration-resistant prostate cancer [82]. Patients were defined to have castration-resistant prostate cancer and eligible for the study, after anti-androgen therapy had been stopped for at least 4 weeks before the trial and serum PSA levels were at least 5 μg/L having increased 3 successive times, 2 weeks apart in the presence of castrate levels of serum testosterone. Sodium selenate was administered daily for 3 weeks. Initially, the sodium selenate was given at a fixed dose (one patient each at 5, 10, 15 and 30 mg), however, after observing the short half-life in serum, the same total daily dose was administered in three separate doses throughout the day in order to generate steady state plasma levels of sodium selenate. During the two years of enrollment, 19 patients were enrolled with a mean age of 72. 12 of these patients completed the treatment for 12 weeks. Of the other 7, 4 withdrew from the study due to disease progression, 1 with grade 3 fatigue, another with concomitant grade 3 diarrhea, muscle cramps and acute renal impairment. These three patients were all in the 90 mg dose group, receiving 30 mg sodium selenate trice daily. The only serious adverse event that could have been due to sodium selenate was in the patient that experienced acute renal impairment (increased creatinine level from 90 mmol/L to 260 mmol/L). From the patient’s medical records, it was noted that they had a prior history of underlying kidney disease, even though the immediate cause could not be determined. Therefore, sodium selenate’s role in this occurrence could not be ruled out.
Due to the short half-life in the single dose treatments, the recommended phase II dose of 20 mg trice daily, showed a half-life of 2.9 hours and Tmax of 2.5 hours. Selenite was the major metabolite in the plasma and reached steady-state levels by three weeks. On the other hand, while selenite was hardly detected in the urine after 24 hours, selenate, SeMet and other methyl selenium species were the major selenium compounds identified. As a surrogate marker of tumor progression, PSA was monitored throughout the study. One patient had a 57% reduction in PSA and two patients’ tumor stabilized for 28 and 41 weeks. For all other patients who completed the 12-week scheduled treatment, the mean doubling time of PSA increased from 2.2 to 4.0 months. As this study was not designed in order to assess the efficacy of sodium selenate, its probable role in the treatment of CRPC and other cancers should be further examined. It is not surprising that humans tolerated super-high doses of selenate (compared to 200 ug SeMet used in SELECT and other trials) when considering that selenate was almost biologically inert in cell culture models.
The FDA approval of Investigational New Drug IND status for MM-Se is required for human studies in the US. Preclinical short and long term toxicology studies of MSeC have been conducted by the National Cancer Institute through the Division of Cancer Prevention (DCP) Rapid Access to Preventive Intervention Development (RAPID) Program [83]. Male and female CD rats received daily gavage doses of 0, 0.5, 1.0, or 2.0 mg Se/kg/day (0, 3, 6, or 12 mg/m2/day). Also, both male and female beagle dogs received daily gavage doses of 0, 0.15, 0.3, or 0.6 mg Se/kg/day (0, 3, 6, or 12 mg/m2/day) for 28 days. In the rats, MSeC induced dose-related hepatomegaly in both sexes; mild anemia, thrombocytopenia, and elevated liver enzymes were observed in high dose females only. In the middle and high dose females, there was a statistically significant observed decrease in hematocrit, hemoglobin, mean RBC volume and platelet count. Microscopic pathology included hepatocellular degeneration (high dose males, all doses in females), arrested spermatogenesis (high dose males), and atrophy of corpora lutea (middle and high dose females). In dogs, MSeC induced mild anemia in middle and high dose males, and in high dose females. Reduced hematocrit and RBC count were observed in the middle and high dose dogs and reduced hemoglobin in only the high dose males. Toxicologically significant microscopic lesions in dogs were seen only in the liver (peliosis and vacuolar degeneration in high dose males, midzonal necrosis in males in all dose groups). Based on liver pathology seen in female rats in all dose groups, the no observed adverse effect level (NOAEL) for MSeC in rats is <0.5 mg Se/kg/day. Based on alterations in hematology parameters and liver morphology in male dogs in all dose groups, the NOAEL for MSeC in dogs is < 0.15 mg Se/kg/day. Taking the NOAEL of dogs as reference (~ 0.1 mg/kg), an equivalent value extrapolated to humans for a 70 kg person is 7,000 μg Se/day.
Roswell Park investigators led by J. Marshall conducted and published the first-in-human single-dose pharmacokinetics of MSeC in men [84]. In this randomized and double-blinded study, subjects received either a single dose of MSeC at 1 of 3 different concentrations or the placebo. In the first wave, 5 subjects received 400 μg of Se and 1 received placebo; in the second wave, 5 subjects received 800 μg of Se and 1 received placebo; in the third wave, 5 subjects received 1,200 μg of Se and 1 received placebo. The results show that the most distinct concentration curve is for the 1,200 μg dose, although the curve of the 800 μg dose slightly exceeds that of the 400 μg dose and that of the 400 μg dose exceeds that of placebo. For those receiving Se, Tmax are similar, ranging between 3 and 5 hours for the 400 through 1,200 μg cohorts. The mean Se Cmax increases in a dose–response fashion from 10 for placebo to 22.8, 30.75, and 63.2 ng/mL (~0.8 μM) for 400, 800, and 1,200 μg dose subjects, respectively. There were 25 grade 1 adverse events reported, with no association between the administration or dose of MSeC to the adverse event experienced. As a result, no evidence of toxicity could be established. Similar studies should be planned and conducted for MSeA given the significant differences in rodent proteomic profiles of these two MM-Se highlighted earlier [59,85].
Mechanistic studies have indicated that the forms of Se are critical for therapy, depending on their entry into two distinct Se metabolite pools that exert diverse and differential effects on signaling pathways, leading to proliferative arrest and cell death/apoptosis. In cell culture models, MM-Se (the methylselenol-MSeA redox duet) have many desirable attributes of cancer chemoprevention and therapy, including targeting key signaling pathways, angiogenic switch regulators, invasion and metastasis molecules and in general cancers as well as sex hormone signaling in gender-specific cancers. The hydrogen seide pool in excess of selenoprotein synthesis can lead to DNA single strand breaks and genotoxicity to normal cells. In several animal models, the MM-Se compounds used alone exhibited greater (adjuvant) therapeutic efficacy in treating several cancer types, for example breast and prostate cancer models than SeMet. Their utility as chemoenhancer by improving the efficacy of approved drug modalities is promising and more preclinical animal efficacy studies are warranted. Several potential mechanisms include suppression of prosurvival molecules and increased caspase activation, enhanced drug uptake and retention, and decreased toxicity of chemodrugs to host (Figure 2). Accumulating data support MSeC and MSeA as more meritorious candidates than SeMet or selenite for future clinical investigations of cancer therapeutic efficacy, especially in the combination with chemotherapies, and perhaps biotherapies and radiotherapy. Our proteomic analyses of prostate and their lesions from MSeA vs. MSeC-treated mice indicated these forms are non-exchangeable. Therefore, their safety and efficacy should be each rigorously studied and compared in animal models, preferably higher mammals than rodents, to provide solid scientific choice of a preferable form for human translation.
We regret the inability to cite many worthy papers due to space limitations. We thank Jinhui Zhang, PhD and Michael Melkus, PhD for help and valuable contributions. This work was supported by US National Cancer Institute R01 grant CA172169.