Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2017) Volume 10, Issue 8
The capability of molecular docking in drug discovery can never be underestimated. The success of number of FDA approved drugs for several dreadful diseases have enhanced the speed of drug discovery. Although the inconsistent track record of computational screening may increase the doubts that how well the docking methods can rank the New Chemical Entity. If the method is studied correctly the docking method can have capability to screen and rank a true ligand from false ligand.
Here, the performance of molecular docking studies has been evaluated by correlations of experimental binding affinities of 3D ligand-enzyme complexes of Bcr-Abl. Here we evaluate the effect of the protein-ligand complex with the different scoring function and explained how to screen the analogs in better way by using existing computational approach.
This review and computational work will certainly boost the confidence of computational biologists.
Keywords: Molecular docking; NCE; Scoring analysis; PLP1; PLP2
The efforts of computational biologist are frequently being questioned, that how well the docking works. The question becomes more prominent by some inconsistent records. If we look at the success story, we can tell that the computational biologists are making an effort to understand the diseases with just a “MOUSE CLICK”. Their effort have already resulted in the success of Imatinib, Ponatinib, Vemurafenib etc are few among the list. Molecular docking is a method of prediction of techniques that, how small molecules fit in binding sites fit and interacts with the binding sites residues in a protein complex.
Although the number of literatures and drug discovery approaches has given adequate answers to the above questions, still we have made an attempt to study similar compounds for BCR- Abl T315 reported in Bio-organic and Medicinal chemistry [1-3] to answer the question in favor of computational biologist. The docking method should predict the different poses of a ligand inside the binding pocket of a protein. If correct binding pose is not selected we may end up wrongly in ranking of NCEs. When we looked back the history of last 20 years, we have found that how rarely sufficient evidence is present to rank the NCEs.
Generally the screening based on scoring analysis and top scoring hits are investigated by investigators. Those investigators are known as computational biologists. Here comes the knowledge of a computational biologist, whose job is just screen and ranks the molecules with the probability of its activity in human. The method of docking screen different poses of the molecule. The pose selection is very important as the docking scores are dependent on these.
The prediction of correct ligand pose or orientation is one of the most important things in ranking of NCEs. This depends on how well computational docking program predict the binding pocket. This can be done by comparison of the crystal structure pose to the docked pose.
Structure based virtual screening has become integral part of drug discovery. There are number of types scoring values available for screening the molecules such as LIGscore, Dock Score, PMF, PLP, and PLP2.
The screening of analogues based on scoring values is still question of debate among the computational biologists & medicinal Chemistry groups. Several questions arise in mind such that which scoring value should be used for screening the analogues.
In this study we aimed to perform the evaluation of some of available scoring functions in Discovery Studio. Success of molecular docking depends on how the ligand binds to the defined binding site of a protein. In our paper we have studied how to analyse the different scoring functions for screening of a molecules.
The first step is retrieving of the required protein structure from PDB. The downloaded protein is then subjected for proteinpreparation. The protein- preparation tools manipulate and investigate protein structures.
Protein preparation
The 3D structures of PI3K were downloaded from PDB (Table 1).
| Molecule | R1 | R2 |
|---|---|---|
| Ponatinib | 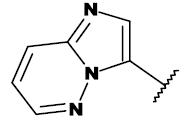 |
CH3 |
| 7c | 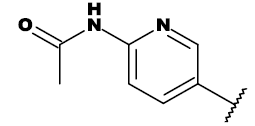 |
CH3 |
| 7f | 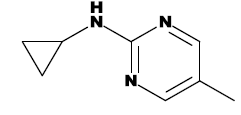 |
CH3 |
| 8e | 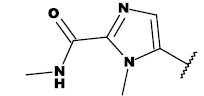 |
CH3 |
| 8h | 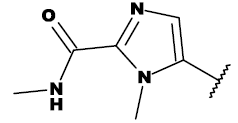 |
CH2OH |
| 19a | 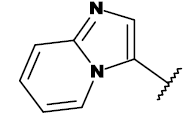 |
CH3 |
| 20a | 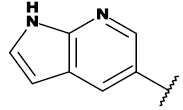 |
CH3 |
| 20c | 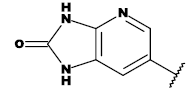 |
CH3 |
| 20i |  |
CH2OH |
Table 1: List of structures taken for molecular docking studies.
The downloaded PDB ID 3IK3 was subjected for protein-preparations. Water molecules apart from 3 Angs were removed. Hydrogens were added to protein-ligand complex. Prepare protein protocol of Discovery studio was used for preparing protein. Missing residues were added.
• PDB ID: 3IK3.
• Type of enzyme: BCR-ABL.
• Resolution Angstrom: 1.9.
• IC50 (nM): 2.
Analysis of binding sites
The binding site of PDB ID 3IK3 is analyzed for hydrogen bonds and the presence of surrounding residues.
Ligand preparation
The ligands for target BCR-ABL were taken from Bio Organic Medicinal Chemistry articles [3]. All these ligand have reported IC50 Values. The set of ligand were drawn and minimized using discovery studio prepares Ligand protocols. The structure list is given in Table 2 [3,4].
| Molecule | Scoring Value | ||||||
|---|---|---|---|---|---|---|---|
| -PLP1 | -PLP2 | LigScore1 | LigScore2 | D-Score | PMF | IC50 (nm) | |
| Ponatinib (Ref) | 179.54 | 162.17 | 7.32 | 8.7 | 151.406 | 204.28 | 11 |
| 7b | 161.3 | 143.5 | 7 | 8.41 | 145.137 | 176.33 | 65 |
| 7c | 184 | 165.03 | 7.95 | 8.92 | 156.148 | 202.14 | 9.6 |
| 7f | 184.55 | 165.95 | 7.8 | 8.76 | 170.521 | 198.52 | 519 |
| 8a | 155.05 | 138.23 | 6.86 | 8.24 | 130.027 | 167.78 | 5738 |
| 8b | 163.07 | 146.62 | 7.01 | 8.45 | 138.952 | 175.79 | 709 |
| 8e | 185.18 | 165.99 | 7.69 | 8.71 | 155.921 | 191.06 | 4 |
| 8h | 195.68 | 181.82 | 8.82 | 9.15 | 140.008 | 193.47 | 8.4 |
| 12a | 170.69 | 154.01 | 7 | 7.81 | 143.608 | 184.76 | 72 |
| 12c | 164 | 147.97 | 6.4 | 7.73 | 143.631 | 183.32 | 471 |
| 19a | 179.44 | 162.6 | 7.27 | 8.73 | 148.73 | 205.94 | 26 |
| 19d | 177.35 | 160.69 | 7.16 | 8.56 | 154.56 | 206.83 | 727 |
| 19h | 181.67 | 164.36 | 7.32 | 8.75 | 156.338 | 209.47 | 49 |
| 20a | 182.42 | 163.46 | 7.87 | 9.07 | 149.373 | 204.5 | 14 |
| 20b | 175.19 | 157.94 | 7.24 | 8.7 | 143.515 | 199.57 | 42 |
| 20c | 185.63 | 165.7 | 8.15 | 8.96 | 165.869 | 191.01 | 31 |
| 20d | 187.4 | 167.9 | 8 | 8.9 | 143.948 | 199.34 | 17 |
| 20e | 178.88 | 160.55 | 7.2 | 8.7 | 135.809 | 202.28 | 500 |
| 20f | 178.55 | 160.14 | 7.24 | 8.67 | 155.024 | 196.48 | 13 |
| 20i | 182.19 | 164.47 | 7.88 | 8.93 | 154.848 | 208.3 | 14 |
| 20j | 176.22 | 162.27 | 7.22 | 8.57 | 153.96 | 200.6 | 34 |
| 20p | 159.12 | 149.63 | 6.6 | 8.05 | 146.446 | 169.07 | 1132 |
| 24a | 175.84 | 157.16 | 7.02 | 8.48 | 151.074 | 218.97 | 295 |
Table 2: Scoring table (LigandFIT).
Molecular docking studies
The Ligand FIT program of Discovery Studio was used to screen small set of ligands. The LigandFit approach is shape based algorithm. The docking process consists of two major parts. The first step is specification of binding site region of receptor to use for docking studies. The second step is docking of ligands to specified site. The binding site will be further evaluated by different scoring analysis.
Validation of developed docking parameter
This step involves comparison of crystal pose with docked pose. The RMSD should not be more than 1 Angs.
This analysis is an integral part of docking as it is very helpful we are ranking the NCEs. The analysis of PDB ID 3IK3 reveals the presence of four hydrogen bonds.
The binding site analysis of Ponatinib indicates that the imadiazole core rests in adenine pocket of enzyme. The methyl phenyl group occupies a hydrophobic pocket behind I315, the ethynyl linkage forms favorable van der Waals interactions with the amino acid and the trifluoromethyl group binds to a pocket induced by the inactive conformation kinase.
Ponatinib avoids the steric clash with the bulky side chain of gatekeeper residue isoleucine 315, with a linear triple bond linkage as given in Figure 1. By bypassing the original steric hindrance introduced by isoleucine, Ponatinib is able to interact with the hydrophobic pocket while maintaining favorable hydrogen bonding interactions, including interactions with residues Glu 286, Met 318, His 361, and Ile 360 on the WT and mutant Bcr-Abl (Figure 2).

Figure 1: Isoleucine 315, with a linear triple bond linkage.

Figure 2: Binding site analysis of Ponatinib.
The ligand is surrounded by hydrophobic residues. The substituted groups of reported 22 analogs are analyzed based on surrounding residues.
The comparison of docked Ponatinib structure with docked pose is given below in Figure 3. The RMSD between two is less than 1 Angstrom.

Figure 3: Superimposition of Docked pose (grey) with crystal structure (yellow).
The same developed docking parameter was used for docking the analogs.
PLP1 and PLP2 scoring values are developed by Gehlaar et al. [2]. PLP excludes hydrogen atom from calculation. The four atom number considered by PLP are Hydrogen bond donor, Hydrogen Bond acceptor only, both hydrogen bond and acceptor and Non-polar.
Based on different scoring values PLP1, PLP2, LigScore-1, LigScore-2, D-score and PMF, we have selected 7c, 7f, 8e, 8h, 19a, 19h, 20a, 20c and 20i. The scoring values are given in Table 2. When we have analyzed the experimental values we have observed that Ic50 of all selected molecules are good except for 7f, which as per reported in article is 519 nM.
Then we have analyzed the binding site and structural differences between Ponatinib and compound 7f. Although PLP1 and PLP2 scoring value are good for this molecules. So we have looked for the reason behind this, we observed that R2 and R-3 is same for Ponatinib and 7f. Only difference lies in R1. Then we looked for R1 changes, which revealed that number of non-polar atom is more in 7f than Ponatinib, which can be the reason for increase in PLP1 and PLP2. LIgscore2 of compound 7F is quite same as Ponatinib as hydrogen bonds are same for both molecules.
Then we have evaluated 7f and Ponatinib, Hydrogen bonds. The Hydrogen is same reported in Figure 4.

Figure 4: (a) H-bonds interaction of 7f, and (4b) H-bonds interaction of Ponatinib.
The numbers of hydrogen bonds are same in both cases. Cyclopropyl ring of compound 7f is lying on the surface. The increase in scoring value of PLP1 and PLP2 can be explained by the increase in number of non-polar atoms. The number of HBD has increased. In case of Ponatinib there are no HBD atoms.
In contrast to Ponatinib, the P-loop of PDB ID 3IK3 7 h making limited van der Waals interactions with the inhibitor. This is most likely caused by the change of the 6,5-fused bicyclic core in Ponatinib compared to the monocyclic core in 7f resulting in the loss of edgeto- face aromatic interactions between Y253 located in the P-loop and the hinge binding core of 7f. Overall, the model suggests that 7f binds similar to Ponatinib in ABLT315I but with the ability forfeiting close van der Waals interactions with the P-loop. The vanderwaal interactions are given in Figures 5 and 6.

Figure 5: The active pocket residues of 7f. Color green represent for van der Waal contacts and color pink represent for electrostatic contacts using with Discovery Studio 3.5 client.

Figure 6: The active pocket residues of Ponatinib. Color green represent for van der Waal contacts and color pink represent for electrostatic contacts using with Discovery Studio 3.5 client. The drug makes intimate van der Waals contacts with Tyr253 due to the kinked or compact conformation of the P-loop and with Phe382 due to the DFG-out mode of the activation loop.
The scoring values reasons were explained clearly. So as a computational biologist we should screen the molecules taking all this in account like how many atoms we have increased or decreased etc.
Then we have tried to look in depth for the reason for compound 7f low in vitro activity. We have found the presence of HBD (Hydrogen bond donor) which is not there in Ponatinib. Number of non-polar atom is also more than Ponatinib. The superimposition structure of compound 7f and Ponatinib is given in Figure 7.

Figure 7: Superimposition structure of Ponatinib (green) and compound 7f.
The modified R1 region is creating adverse effect on the compound 7f. The in vitro activity is reduced from 8 nM to 519 nM.
The compound 7C is have lost the capability to form strong vanderwaals contact as reported for Ponatinib.
The above method has screened the active analogs. The studies have revealed that scoring analysis depends number of changes done in the designed analogs (Figure 8) [5-11].

Figure 8: Figure of compound 7f and Ponatinib (green) along with BCR_ABL protein. The circle region is modified region. As per figure the modified R1- group is lying on the surface without any interactions. This can be reason for low in vitro activity.
We can say that analysis of docking results lie in the hand of investigators. The studies described here have screened the analogs in true ligand and false ligand. The results are approximately matching with experimental data. A computational biologist has to understand that while analysis of results we should not just blindly screen based on scoring analysis. As we see there are a variety of scoring functions available in Discovery Studio for estimation of ligand-receptor interactions. When an atom is changed from Nitrogen to Carbon in the ligand, the atom type (chemistry, bond, hybridization) is changed so should expect different ligand scores. The number of polar atom or non-polar may increase which will result in increase of PLP1 and PLP2.
LigScore2 uses empirical scoring functions based on several descriptors and coefficient parameters. As per LigScore2 function equations; the vdW descriptor term is a soft L-J potential, Nitrogen atom or Carbon atom have different vdW radii therefore different value for this term. Another descriptor is based on the buried polar surface area, which will also be slightly different for the change of N atom to C atom.
The PMF and PMF04 are knowledge-based methods, developed based on statistical analysis of the 3D structures of protein-ligand complexes. The scores are calculated by summing pairwise interaction terms over all interatomic pairs of the receptor-ligand complex. Similarly, Nitrogen or Carbon will have different atom typing parametrization and therefore slightly different PMF scores. So investigators have to understand clearly all these before selecting or ranking the analogs.
These studies have given encouraging idea of screening the NCEs. First and foremost the result analysis is very important in screening. The investigators need this specific knowledge for ranking the molecules.
We must agree the work of computational biologist is not an easy one. The people and process of molecular docking behind Ponatinib, Imatinib, and Vemurafenib etc. must be given their due credit because by just simple MOUSE CLICK system they are giving hopes of life for so many patients. Docking process appears tow work amazingly well with the help of computational biologists. The level of success should comfortably remove our doubts, although we can still improve our process.
At last does docking screen the molecules? Yes, it screens and results in successful drugs.