Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Research Article - (2014) Volume 2, Issue 1
Methicillin-resistant staphylococcus aureus (MRSA) is a bacterium that is evolving towards adaptive changes to certain antibiotics like methicillin, oxacillin, penicillin, and amoxicillin. It is a communicable and most rapidly spreading disease worldwide. It is reported that MRSA is becoming common among children in intensive care units. This disease comes under Emerging Infectious Disease (EID). In this paper, MRSA proteome screening is done and Drug/vaccine targets are proposed based on its essentiality to the pathogen and non-homology with human proteome. Targets validation is done so that its targeting must not affect human proteome and vital pathways. Those targets which has no structure, structure prediction and validation is done and important epitopes and ligands are proposed on suitable targets.
Keywords: MRSA, EPA, Infection, Targets, Epitope, Drug, Pathways
MRSA spreads infection through blood and skin. The former infection is of severe kind. But skin infections are the most common one. Skin infection appears as pustules or boils which generally form at areas of the body covered by hair like back of neck, groin, buttock, armpit, beard area of men. The degree of symptoms depends on the stage of infection. The persons who meet MRSA patients are at Risk of Acquiring MRSA Infections. In hospitals, patients has more risk of getting MRSA through catheters inserted into the skin and those patients who has undergo medical procedures like surgery. According to drug bank only three antibiotics namely Arbekacin, Meticillin and Linezolid, are approved for the treatment of MRSA. Drug targets are limited and there is an urgent need for the discovery of novel drug targets. Apart from drug target, we also need Good ligand formulation that would act as a potent inhibitors to the novel targets without affecting human proteome (Figure 1).

Figure 1: MRSA.
Any contaminated surfaces is the source of many kind of infections, MRSA is one of them. Mishandled Hospital procedures can leave immune compromised patients vulnerable to MRSA. Environmental hygienic conditions are the source of good physical and mental health. It is reported that U.S. Environmental Protection Agency (EPA) labeled Cleaners, disinfectants and sanitizers must be used in order to spread all kinds of infections.
Staphylococcus aureus proteome screening
Prokaryotic sequence homology analysis tool (PSAT) [1] is used for finding conserved patches in all strains of Staphylococcus aureus and Streptococcus, as both are heavily involved in skin infections. Yersinia Pestis CO 92 is taken as reference strain. Following parameters is considered for finding conserved genes in these selected pathogens: BLAST alignment score thresholds for finding gene homologs: e-value < 10; bit score > 20; % identity > 10
DEG BLAST
All the conserved genes are checked for their key role to pathogens. Database of essential genes (DEG) [2] is used to screen all the conserved genes for their vitality to the selected pathogens. This tool gives hit if query gene is showing any significant similarity with pathogen’s growth, reproduction and survival.
NCBI Homo sapiens (human) protein BLAST
Those conserved genes which are giving hits in DEG blast were further checked in NCBI Homo sapiens (human) Protein BLAST [3]. If we are proposing target on pathogen proteome then those conserved genes must be checked if there is any similarity with human proteome because drug/vaccine targeting must not affect any of the human protein.
Annotation and pathways analysis
The conserved genes which are showing no significant similarity checked in NCBI Homo sapiens (human) Protein BLAST was checked for annotation and their involvement in any crucial pathway. Annotation was done by using CELLO [4] and PSORTB [5]. Pathway analysis was done in KEGG genes database [6].
Target proposal and structure prediction
Based on the pathway study, its essentiality and non-homolog to human proteome property of Targets, they are proposed for drug targets and epitope design. Those targets whose have no protein structure was modeled by Protein Homology/analogy Recognition Engine (PHYRE) [7].
Structure validation
These Target proteins were optimized from KOBAMIN [8] and Galaxy WEB server [9] and validated in Rampage [10] and Erratplot [11], the former check the stereo chemical properties of modeled structures and latter one analyze the non-bonded interactions.
Epitope design
Transmembrane region of proposed target was predicted with TMHMM [12]. After that, Prediction of B cell epitopes with BCpred (cutoff 0.8, 20-mer epitopes) [13]. Generally B cell epitope sequences are surface-exposed of corresponding proteins. T cell epitopes from propred [14], propred 1[15], MHCpred [16] and T epitope designer [17]. Those T cell epitopes were considered that is part of B cell epitopes and lies at Transmembrane region according to TMHMM. Predict those epitope's antigenicity from vaxijen [18]. Finally, epitopes that bound more than 13 MHC molecules in ProPred and ProPred-I with less than 100 nM IC50 for DRB1*0101 in MHCPred v2.0 and that bound >=80% of HLA molecules in T-epitope designer were selected. Proteins that were antigenic according to Vaxijen (threshold=0.4, ACC output) score above than 0.5 in VaxiJen were selected.
Virtual screening of ligands against target proteins
All the target proteins were screened with 12 million drug-like ZINC12 + 6507 DrugBank drugs from FINDSITE-COMB [19]. Less characterized ZINC DATABASE molecules were selected for druglikeness analysis from FAF- Drugs2 [20]. FAF results were cross checked with Osiris property explorer [21]. We modified most of the ZINC ID ligands to satify druglikeness and ADME properties. We proposed those novel ligands for each target which is following all the drug-likeness properties.
Molecular docking studies of ADMET following ligands with target proteins
ADMET following ligands were docked with their corresponding target in particular coordinate predicted by Pocket-Finder [22] using Molegro Virtual Docker [23]. All the docking parameters and images were finalized for each ligand-protein docking.
Six targets are finalized for further study (Table 1). Protein accession number, locus tags are the unique identifier of insilico discovered targets. All are essential for pathogens as inferred from DEG Blast results. kdpA, opp-1B and icac structures are not available. Therefore, they must be modeled for further molecular docking studies. Pathways and Cellular localization results are shown in Table 2.
| Locus Tag | Protein ID | Target name | Structure/ Related (yes/no) | DEG hits (yes/no) |
|---|---|---|---|---|
| SAR2165 | YP_041527.1 | kdpA | no | yes |
| SAR2553 | YP_041904.1 | Opp-1B | no | yes |
| SAR2750 | YP_042088.1 | icac | no | yes |
| SAR0118 | YP_039582.1 | sirA | Yes (Related: pdbid: 3MWF) | yes |
| SAR2537 | YP_041888.1 | opuCB | Yes (Related: pdbid: 3D31 C) | yes |
| SAS0639 | YP_042767.1 | Hypothetical protein | Yes (Related: pdbid: 3MLV L) | yes |
Table 1: Final Targets based on subtractive proteome screening.
| Target name | CELLO | PSORTB | Pathway |
|---|---|---|---|
| kdpA | Membrane | Cytoplasmic membrane | Two-component system |
| Opp-1B | Membrane | Cytoplasmic membrane | ABC transporters |
| icac | Membrane | Cytoplasmic membrane | No pathway |
| sirA | Periplasmic | Cytoplasmic membrane | ABC transporters |
| opuCB | Inner membrane | Cytoplasmic membrane | ABC transporters |
| Hypothetical protein | Membrane | Cytoplasmic membrane | No pathway |
Table 2: Cellular localization and pathways of target proteins.
All the target proteins are suitable for epitope design as all are predicted to be localized in Membrane from CELLO and PSORTB. They can also be considered for potent drug target. Function of Target Protein is very important for Target validation. Functional annotation is done in Table 3. Epitope design on the target proteins are given in Table 4. The final epitopes following all the criteria from target proteins are given in Table 5. The structural information of refined modeled target proteins from erratplot and rampage are shown in Tables 6 and 7. The coordinates of best pockets for each target proteins are given in Table 8.
| Target name | Function | Molecular Function |
|---|---|---|
| kdpA | One of the components of the high-affinity ATP-driven potassium transport (or KDP) system, which catalyzes the hydrolysis of ATP coupled with the exchange of hydrogen and potassium ions | ATP binding, potassium-transporting ATPase activity |
| Opp-1B | Oligopeptide transporter putative membrane permease domain | transporter activity |
| icac | Presumably involved in the export of the biofilm adhesin polysaccharide poly-beta-1,6-N-acetyl-D-glucosamine (PNAG, also referred to as PIA) across the cell membrane | transferase activity, transferring acyl groups other than amino-acyl groups |
| sirA | Iron-regulated ABC transporter siderophore-binding protein SirA | Iron-regulated ABC transporter siderophore-binding protein SirA |
| opuCB | Probable glycine betaine/carnitine/choline ABC transporter opuCB | transporter activity |
| Hypothetical protein | Uncharacterized protein conserved in bacteria [Function unknown] | Function unknown |
Table 3: Function annotation of target proteins.
| Target name | TMHMM | Bcpred Position-Bcell epitope (confidance value) |
Propred 1 (Sorted in descending score) |
Propred | MHCpred |
|---|---|---|---|---|---|
| kdpA | Outside: 1-3 86-126 192-244 303-324 373-375 438-483 549-558 TMhelix: 4-26 63-85 127-149 169-191 245-267 280-302 325-347 354-372 376-398 415-437 484-506 526-548 |
462-AAANNGSGFEGLKDDTTFWN (0.97) 335- FTVITTAFTTGSVNNMHDSL (0.92) 77-LLIVQQWLFLNPNHNLNQSI(0.91) 434 AFMIPGASESITNPSFHGIS (0.89) |
LIVQQWLFL NNGSGFEGL MIPGASESI AAANNGSGF LLIVQQWLF IPGASESIT FTVITTAFT TVITTAFTT FMIPGASES ITTAFTTGS VITTAFTTG ANNGSGFEG IVQQWLFLN |
FMIPGASES (10/51) | FMIPGASES - 72% MHC alleles are Binding. |
| Opp-1B | outside 32-106 162-175 257-275 TMhelix: 9-31 107-126 139-161 176-193 234-256 276-298 |
80- NFGTSYITGDPVAERIGPAF (0.99) 41- AQGTPNVTPELIAETNEKYG (0.91) | GTPNNTPEL TPNVTPELI TSYITGDPV GTSYITGDP AQGTPNVTP FGTSYITGD NFGTSYITG QGTPNVTPE |
YITGDPV (8/51) | YITGDPV -100% MHC alleles are Binding. |
| Icac | outside 30-43 102-115 168-186 234-242 292-305 Tmhelix: 7-29 44-66 79-101 116-138 145-167 187-204 211-233 243-262 269-291 306-328 |
165- YFTNNTAFHDTVLHYYPLSE (0.7) | NNTAFHDTV FTNNTAFHD TNNTAFHDT |
YFTNNTAFH (14/51) VLHYYPLSE (8/51) | FTNNTAFH 100% MHC alleles are Binding. |
| sirA | Outside: Whole protein |
20-GCSGNSNKQSSDSKDKETTS (0.99) 70-LGVKPVGAVESWTQKPKFEY (0.99) 149-KDTTKLMGKALGKEKEAEDL (0.97) 269-LVKKTESEWTSSKEWKNLDA (0.97) 43-AMGTTEIKGKPKRVVTLYQG (0.96) 91-KNDLKDTKIVGQEPAPNLEE (0.94) 177-AAFQKDAKAKYKDAWPLKAS (0.84) |
KNDLKDTKI TTKLMGKAL FQKDAKAKY LGVKPVGAV AAFQKDAKA KPVGGAVESW NDLKDTKIV TTEIKGKPK KDTTKLMGK KKTESEWTS LVKKTESEW NDLKDTKIV CSGGNSNKQS KTESEWTSS GCSGGNSNKQ SGNSNNQSS DTTKLMGKA MGTTEIKGKP AFQKDAKAK GVKPGAVE DLKDTKIVG VKPAVES AMGTTEIKG GTTEIKGKP QKDAKAKYK VKKTESEWT GNSNKQSSD TKLMGKALG |
LGVKPVGAV (4/51) VKPVGAVES (10/51) FQKDAKAKY (8/51) | LGVKPVGAV 68% MHC alleles are Binding. VKPVGAVES 72% MHC alleles are Binding. FQKDAKAKY 80% MHC alleles are Binding. |
| opuCB | TMHelix 114 135 115 134 144 170 145 168 174 189 175 188 223 248 225 235 238 243 268 292 269 290 |
155- YVGAGGLGDFIFNGLNLYDP (0.9) | YVGAGGLGD VGAGGLGDF AGGLGDFIF GAGGLGDFI |
YVGAGGLGD (8/51) | YVGAGGLGD 72% MHC alleles are Binding. |
| Hypothetical protein | TMHelix 64 102 65 102 113 131 115 129 150 181 151 180 194 222 195 202 205 221 |
No B cell epitope predicted. | No common T cell epitope | No common T cell epitope | No epitope binding affinity prediction. |
Table 4: Epitope designing on target proteins.
| Target name | Final Predicted epitopes | Vaxijen score (>0.5) |
|---|---|---|
| kdpA | FMIPGASES | 0.4000 |
| Opp-1B | YITGDPV | 1.2802 |
| icac | FTNNTAFH | 0.3954 |
| sirA | LGVKPVGAV VKPVGAVES FQKDAKAKY | 0.0443 0.0286 1.9244 |
| opuCB | YVGAGGLGD | 1.5458 |
| Hypothetical protein | No predicted epitope | No predicted epitope |
Note: red color indicates non promising peptides whereas green color indicates promising one.
Table 5: Final Epitopes on target proteins.
| Target Name/Protein ID | Ramachandran parameters | Errat plot quality factor | |
|---|---|---|---|
| kdpA/ YP_041527.1 | RMSD | 2.290 | 78.46 |
| Clash score | 20.9 | ||
| Poor rotamers |
3.5 | ||
| Rama favored | 90.6 | ||
| Opp-1B/ YP_041904.1 | RMSD | 8.941 | 82.7 |
| Clash score | 15.7 | ||
| Poor rotamers |
0.4 | ||
| Rama favored | 94.2 | ||
| Icac/ YP_042088.1 | RMSD | 5.597 | 93.59 |
| Clash score | 17.4 | ||
| Poor rotamers |
2.2 | ||
| Rama favored | 95.7 | ||
| sirA/ YP_039582.1 | RMSD | 4.377 | 86.00 |
| Clash score | 8.6 | ||
| Poor rotamers |
1.0 | ||
| Rama favored | 98.2 | ||
| opuCB/ YP_041888.1 | RMSD | 5.944 | 94.44 |
| Clash score | 10.9 | ||
| Poor rotamers |
1.2 | ||
| Rama favored | 96.2 | ||
| Hypothetical protein/ YP_042767.1 | RMSD | 0.749 | 77.03 |
| Clash score | 17.0 | ||
| Poor rotamers |
1.0 | ||
| Rama favored | 91.2 | ||
Table 6: Structural information of target proteins.
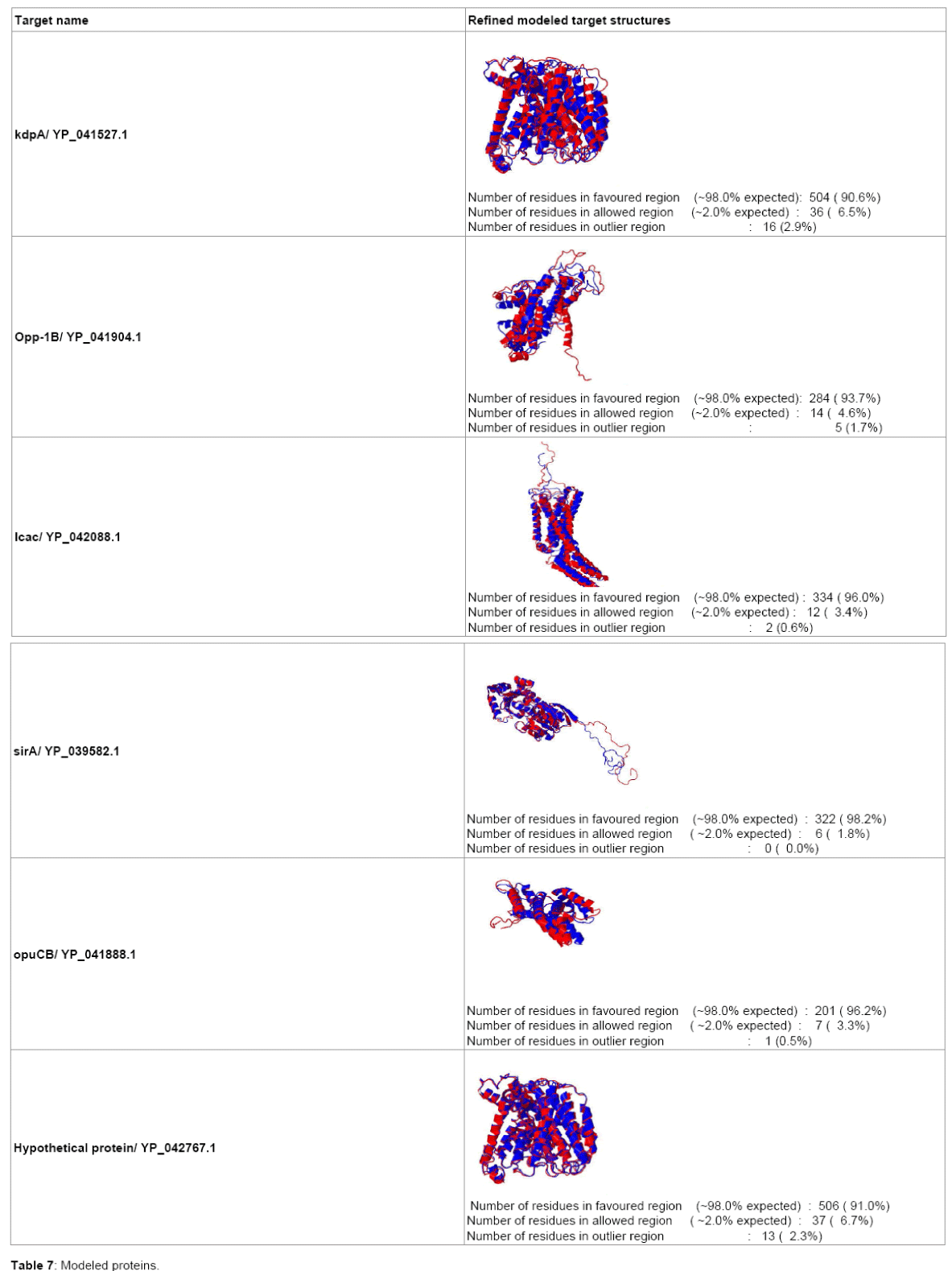
Table 7: Modeled proteins.
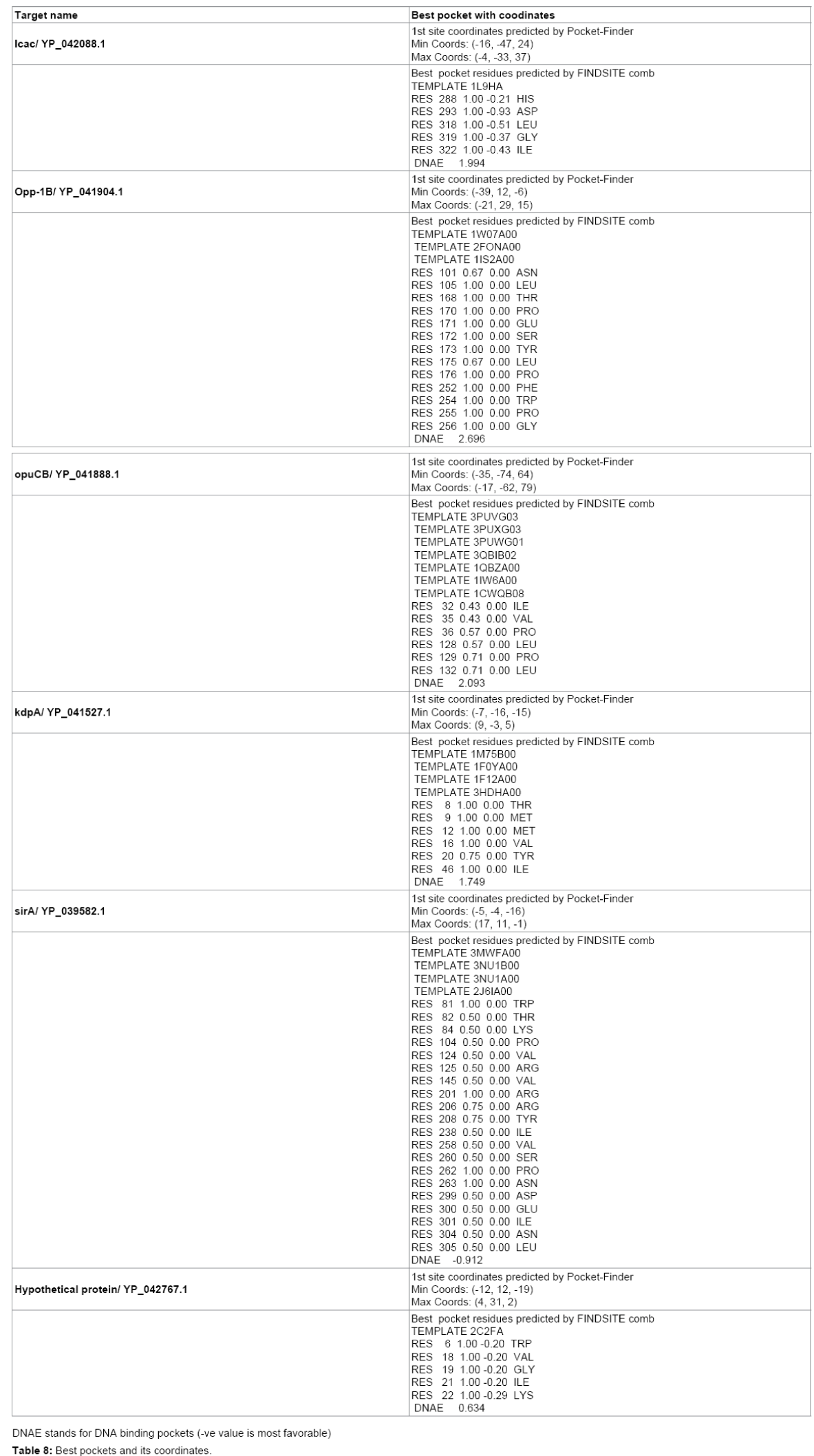
Table 8: Best pockets and its coordinates.