Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2015) Volume 4, Issue 3
HCV NS3 protease domain has been attractive site for inhibition by several direct-acting antiviral drugs. A great success was achieved in treatment of HCV genotype 1 but HCV genotype 4a dominant in Egypt is resistant for many of these drugs while sensitive to some and the causes of these observations have not been deeply investigated. So we constructed a 3D model of HCV NS3 of genotype 4a using HCV NS3 genotype 1b as a template PDB (1DY9) and after close inspection of differences between the model and the template that would alter the drug susceptibility in HCV genotype 4a we performed a comparative computational docking study of Simeprevir, Vaniprevir, and Paritaprevir in NS3 protease domain of both the genotypes. The result of our study successfully explains the difference in response to treatment by HCV NS3 protease inhibitors drugs for both genotypes. It shows that Simeprevir retains its activity in both genotypes but Paritaprevir loses a significant part of its activity that cannot be used alone in HCV genotype 4a, while Vaniprevir remarkably loses its activity that cannot be used in HCV genotype 4a at all. Dynamic simulation of the 3D model of HCV NS3 of genotype 4a was done to augment the docking result. Then a series of some modified inhibitors were virtually screened against our model and this would open a new era to use structure based drug design in developing new drugs that act preferentially in treatment of HCV genotype 4a or other genotypes dominant in developing countries.
<Keywords: HCV; NS3/4A; Genotype 1b; Genotype 4a homology modeling; Docking; Dynamic simulation
Hepatitis C Virus (HCV) is a global health concern that is chronically affecting approximately 200 million people worldwide, about 3% of world’s population. Those individuals are at high risk to develop cirrhosis, hepatic failure, and hepatocellular carcinoma [1,2]. HCV is an enveloped positive-sense single stranded RNA virus (+ssRNA virus) that belongs to the flaviviridae family with seven main genotypes and many subtypes [3,4]. HCV GT1 is the most common genotype counting about 60% of the global infection. It prevails in America, Europe, and Japan, GT3 predominates in Southeast Asia. Egypt is a country where HCV infection is very severe about 15% of the population are infected with GT4 and the main subtype is GT4a [5-8].The HCV genome polyprotein is cleaved into 11 viral proteins five of them are structural (core protein p21, core protein p19, envelope glycoprotein E1, envelope glycoprotein E2 and core protein p7) and six are non-structural (protease NS2–3, serine protease NS3, non-structural protein 4A, non-structural protein 4B, nonstructural protein 5A, and RNA-directed RNA polymerase) [9,10]. Genotype 1 was paid great attention in developing drugs due to the many isolated crystal structures of its proteins [11-13]. But, variants that are predominant in developing countries have not received much attention [14]. Among the many proteins in HCV replication cycle, the protease domain of NS3 plays a vital role and so it has been targeted for developing direct-acting antiviral agents [15]. The function of NS3 protease is to cleave four downstream sites in the HCV polyprotein that are NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B junctions. The NS3 protease is in many ways a typical -barrel serine protease, with a canonical Asp-His-Ser catalytic triad similar to the well-studied digestive enzymes trypsin and chymotrypsin. It forms a heterodimeric complex with the NS4A protein, an essential cofactor that activates the protease and assists in anchoring the heterodimer to the endoplasmic reticulum [16,17].
The catalytic triad performs general acid-base catalysis on target peptides. In summary, a charge relay system is formed in which the carboxylic group of D81 forms a hydrogen bond with Nd1 of H57 [18]. This event increases the pKa of the histidine side chain from 7 to about 12 [17,19]. Consequently, H57 deprotonates the hydroxyl group of the S139 side chain and a proton shuttles to Ne2 of H57 [18]. The Oc of S139 then nucleophilically attacks the carbonyl carbon of a substrate’s scissile bond resulting in the formation of an oxyanioncontaining tetrahedral intermediate [18,20–22]. At this point, the protonated H57 acts as a general acid assisting in the collapse of the tetrahedral intermediate and the cleavage of the substrate [18,22]. The exerted efforts led to development of many inhibitors of HCV NS3/4A protease, which are in the clinical trials and lead to significant reduction in the viral load of patients [23] and some of them have already been approved including the first two approved direct-oral antiviral Telaprevir and Boceprevir may 2011, Simeprevir Sep 2013, Paritaprevir in Viekira package 2014 and Vaniprevir Aug 2014 by PMDA in Japan. The differential susceptibilities of different drugs to protease variants have been investigated [24-29] but little work has been done to explain this. The approved linear NS3 protease inhibitors Telaprevir and Boceprevir are effective against genotype 1b [30,31] but they are completely ineffective in genotype 4a [32]. However the recently developed NS3 macrocyclic inhibitor Simeprevir has been shown to be effective in genotype 1b and 4a with higher efficacy in genotype 1b [33]. Also Paritaprevir has been shown similar efficacy but with lesser extent toward genotype 4a [34] should it must be used in combination with ritonavir and the NS5A inhibitor Ombitasvir. While Vaniprevir was not effective in genotype 4a and despite the treatment option provided by Simeprevir and Paritaprevir the cost of treatment is very high to be afforded by developing countries as the treatment by Simeprevir for three months costs $66,000 [35,36] and the work done to Develop inhibitors for genotype 4a was very limited due to the lack of 3D structure of genotype 4a proteins. So we constructed our model for NS3/4A protease of genotype 4a using NS3/4A protease of genotype 1b as a template PDB (1DY9) that share 80% identity with the target sequence of genotype 4a. We used our model for docking study to explain the different activity of NS3 protease inhibitors among variable HCV genotypes and to open a new era for developing new inhibitors that act in preference for genotype 4a using structure based drug design. Then we used dynamic simulation to confirm the results.
The sequence for NS3 protease domain of genotype 4a consisting of 174 amino acids was retrieved from other published work [37]. The query sequence was used to identify templates with high identity by using BLAST DS against PDB-nr95 data base. Conditions were optimized to get high identity templates by setting the E-value cutoff 0.0001(the lower the E-value cutoff the higher the identity of resulting templates) the search resulted in NS3 protease of genotype 1b PDB (1DY9) with 80% identity.
Sequence alignment and homology modelling
Sequence alignment was done by threading the GT4a NS3 target sequence on the template PDB (1DY9) sequence using Align Multiple Sequences server which use BLOSUM 62 weight matrix algorithm (alignment score) with gap penalty and extension 11 and 1 respectively. The conserved areas of secondary structure were compared between the two sequences and founded that they share high similarity with minute differences suggesting that a high quality model can be obtained.
Building of the 3D model was done by Modeler using the crystal structure as template. Optimization levels were set to high and loop refinement to true to get high quality model. This result in five models with the best model determined. The energy minimization of the modeled protein was done by using ModRefiner, which follows two-step procedure for constructing full-atom model. The first step builds the backbone for the available C-alpha and performs energy minimization to improve the quality followed by the second step which adds side chain atoms from a rotamer library, and conducts energy minimization to both side chains and backbone conformations [38].
3D structure validation
The final refined NS3 model for HCV GT4a was validated by using PROCHECK (Structural Analysis and Verification Server) to calculate the Ramachandran plot [39]. Verify protein Modeler and Verify protein 3D –profiles servers were used for the model to calculate DOPE (discrete optimization protein energy) and to score the model respectively. Structure superposition and RMSD value calculation in addition to the evaluation of the stereochemistry were also employed for the generated model.
Ligand generation and optimization
The structure of the three drugs (Simeprevir, Paritaprevir and Vaniprevir) was obtained from Pubchem [40] and the drugs were inserted to Discovery Studio 2.5 from their SMILES and the modified structures were drawn on Accelrys Draw 4.2 and inserted from their SMILES to Discovery Studio 2.5. CHARMM force field was applied for energy minimization to obtain a convergence gradient by using CHARMM Boundary Potential Builder.
Docking studies
The active site of NS3/4A is shallow and nonpolar [18]. There are many crystal structures on the PDB available with their inhibitors, some of them were downloaded (4TYD [contains Simeprevir as inhibitor] 3SUD, 3SU4, 3SU3, 3SV6 and 4A92.
Super imposition of the crystal structures was very useful in determining the binding site residues that were found to be Q41, F43, H57, D81, V132, L135, K136, G137, S138, S139, F154, R155, A156, A157 and D168. The modeled NS3 of GT4a and crystal structure of GT1b were uploaded as PDB files to discovery studio 2.5 and the binding site for both the proteins was made by a mean of a cavity surrounding the previously mentioned residues. The minimized proteins with their binding site and the selected drugs and their modifications were used to conduct Comparative molecular docking study between HCV GT4a NS3 and HCV GT1b NS3 with Dock ligands (LibDock), a relatively fast algorithm that conducts ‘Hotspots’ matching of ligand conformation and later docked with Hex to obtain a Receptor-Ligand complex [41] and uses LibDock scoring function for binding energy calculations. High score corresponds to a strong binding and a less score corresponds to a weak or non-existing binding. Comparison was done based on the number of HBond interactions, Pi-Pi interactions, Pi-cation interactions and docking score. The ligand-protein complexes were visualized in discovery studio 2.5 visualization tools.
Methods of dynamic simulation
Dynamic simulation was done by Discovery Studio 2.5 under CHARMM 27 force field for protein [42,43]. At the beginning the 3D structures were solvated using the solvation tool in VMD [44]. The TIP3P model was used for the water molecules [45]. Lengevin dynamics for all no hydrogen atoms with a damping coefficient of 1 ps21 was used in maintaining a constant temperature of 300 K throughout the system. A constant pressure of 1 atm was maintained using a Nose´–Hoover Langevin piston with a period of 100 fs and damping timescale of 50 fs [46]. Periodic boundary conditions were used on a 61 Å cubic box with the long-range electrostatics calculated using the particle mesh Ewald method with a grid point density of 0.92 Å. This process ensured that adjacent copies of the protease were never close enough for shortrange interaction. A cut-off of 12 Å for Vander Waals interactions and a switching distance of 10 Å were used for production runs. The system was simulated for 20ns divided on 2000 step and the average result of each 100 step is saved. The resulted 20 copies of protease were analyzed for the conformation change in G41.
The HCV-4a NS3 protease structure model superposes very well on the threading template structure (1dy9), and the two share 80% sequence identity along with nearly identical structural features. When the two structures superpose, the RMSD in back-bone positions is about 0.21 Å and none of the 174-threaded amino acids fall within the disallowed Ramachandran area and no steric clashes or stereochemical outliers were detected The three catalytic residues H57, D81, and S139 are located in a crevice between the two protease b-barrels as shown in [47–49]. The rigid structures indicate that access to the active site is nearly identical in the structural model HCV-4a and template HCV-1b (Figure 1a).
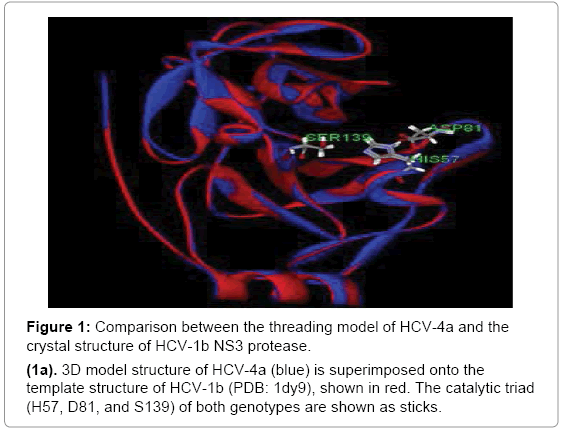
Figure 1: Comparison between the threading model of HCV-4a and the crystal structure of HCV-1b NS3 protease.
(1a). 3D model structure of HCV-4a (blue) is superimposed onto the template structure of HCV-1b (PDB: 1dy9), shown in red. The catalytic triad (H57, D81, and S139) of both genotypes are shown as sticks.
But close inspection of the binding site of two superposed structures with the presence of the inhibitor Vaniprevir for orientation purpose revealed change of some amino acids conformation namely Q41 carboxylic group of GT4a show close proximity towards the cyclopropyl moiety of the inhibitor with distance 1.354 Å leading to steric clash while the distance between the nitrogen of the sulfonyl amide and H57 in GT4a and the GT1b template is 3.6 and 3.0 Å respectively. For GT1b this distance is optimum to form Hbond. between the inhibitor and H57 residue but for GT4a the distance is large enough to diminish the inhibitor`s ability to form Hbond with H57 residue (Figure 1b). These differences in the amino acids conformation suggest the altered binding mode of the inhibitors in both the genotypes and on these basis the docking study was conducted.
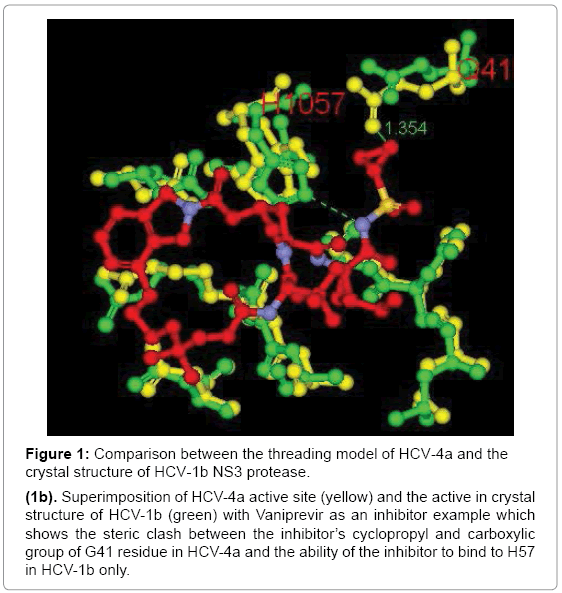
Figure 1: Comparison between the threading model of HCV-4a and the crystal structure of HCV-1b NS3 protease.
(1b). Superimposition of HCV-4a active site (yellow) and the active in crystal structure of HCV-1b (green) with Vaniprevir as an inhibitor example which shows the steric clash between the inhibitor’s cyclopropyl and carboxylic group of G41 residue in HCV-4a and the ability of the inhibitor to bind to H57 in HCV-1b only.
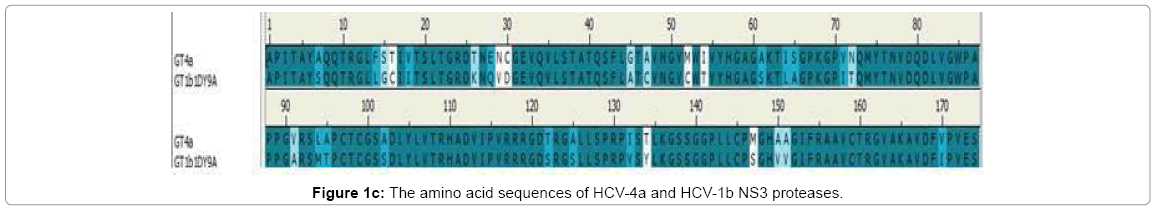
Figure 1c: The amino acid sequences of HCV-4a and HCV-1b NS3 proteases.
Docking studies
First of the beginning docking study was validated by docking of Simeprevir in NS3 of GT1b and comparing the result with Simeprevir in crystal structure PDB (4TYD). In both the docking and crystal structure Simeprevir had the same binding pattern with RMSD ≈ 0.5 (Figure 2a) in general better interaction with the catalytic triad would provide strong inhibition so that the three drugs were docked in NS3 of GT1b and GT4a and the results were compared. In GT1b Simeprevir showed interactions with H57, K136, G137, S139 and R155 while in GT4a it interacts with Q41, H57, K136, G137 and S139 (2Hbonds) (Figure 2b). Proving its ability to treat both the genotypes as it binds to two residues of the catalytic triad (H57 and S139).
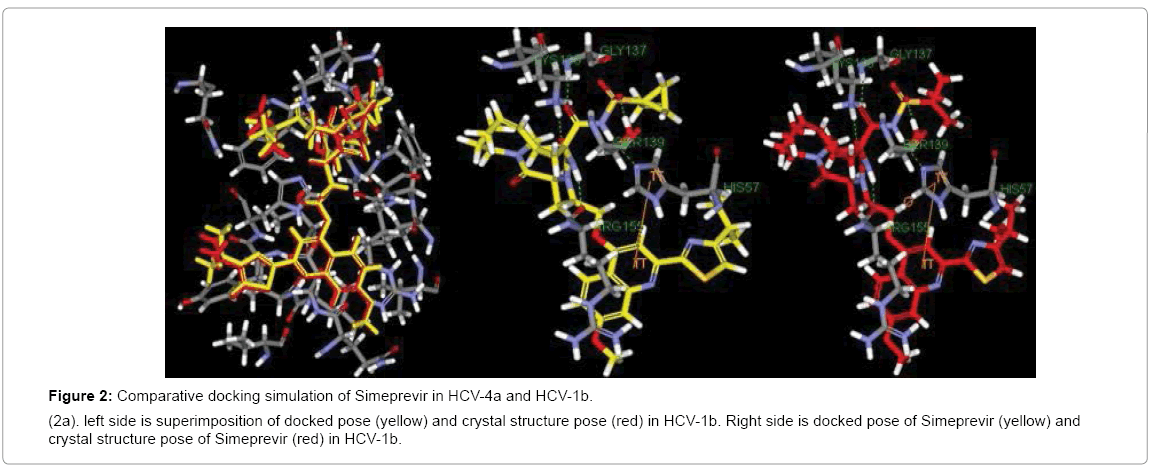
Figure 2a: Comparative docking simulation of Simeprevir in HCV-4a and HCV-1b.
(2a). left side is superimposition of docked pose (yellow) and crystal structure pose (red) in HCV-1b. Right side is docked pose of Simeprevir (yellow) and crystal structure pose of Simeprevir (red) in HCV-1b.
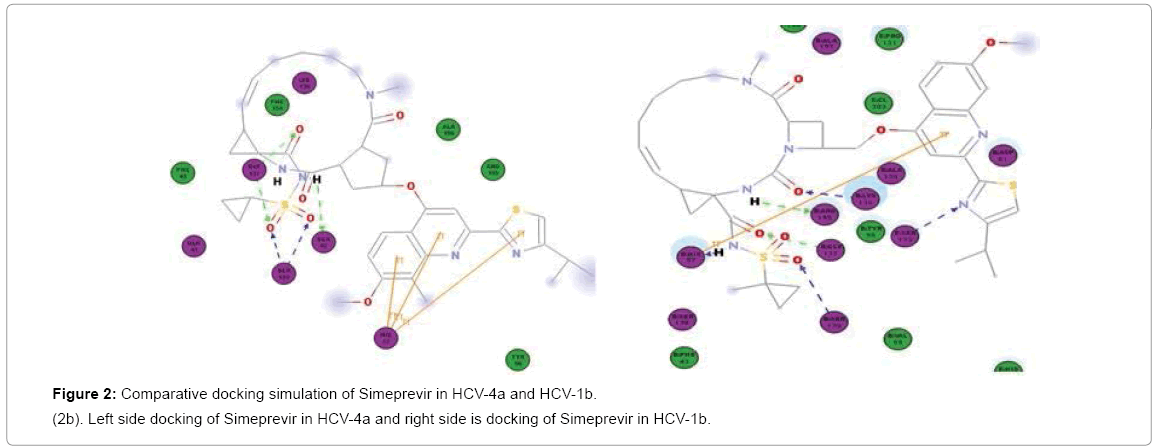
Figure 2b: Comparative docking simulation of Simeprevir in HCV-4a and HCV-1b.
(2b). Left side docking of Simeprevir in HCV-4a and right side is docking of Simeprevir in HCV-1b.
Paritaprevir binds to H57, K136, G137, S139 and R155 in Gt1b but in GT4a it binds well to S139 (3Hbonds) and K136 (3Hbonds) (Figure 3a) So that in GT4a it retain some of its activity but with lesser extent than Simeprevir as it in GT1b it binds to both H57 and S139 while in GT4a it binds to S139 better than GT1b but loses the interaction with H57. Vaniprevir binds to H57, K136 (2Hbonds), S139 and D168 in Gt1b but in GT4a it binds to K136, G137 (2 H-bonds) and S139 (2Hbonds) (Figure 3b) so that it is active in GT1b but not in GT4a as in GT4a it completely loses the interaction with H57 (one of the catalytic triad). So we suggested a series of modification in the cyclopropyl of three inhibitors. The cyclopropyl of the inhibitors was replaced by methyl, bromide, chloride, fluoride or hydroxyl methyl and the same five modifications were done for the three inhibitors and analysis of the docking result of the fifteen compounds in GT4a (sup. information) revealed that the bromide modification of Vaniprevir and Paritaprevir were not capable to interact with H57 and S139 of the HCV NS3 of GT4a because the bromide atom is larger than the cyclopropyl and so maintained the steric clash with Q41 residue and diminished the inhibitors ability to interact with H57 and S139 residues (Figure 4b) but the other modifications were capable of interacting with H57 and S139 and avoiding the clash (sup. Information) because fluoride, chloride, methyl and hydroxyl are smaller than cyclopropyl group of the original inhibitors so that these modifications provide better interaction within the active site of NS3 of GT4a (Figure 4c).
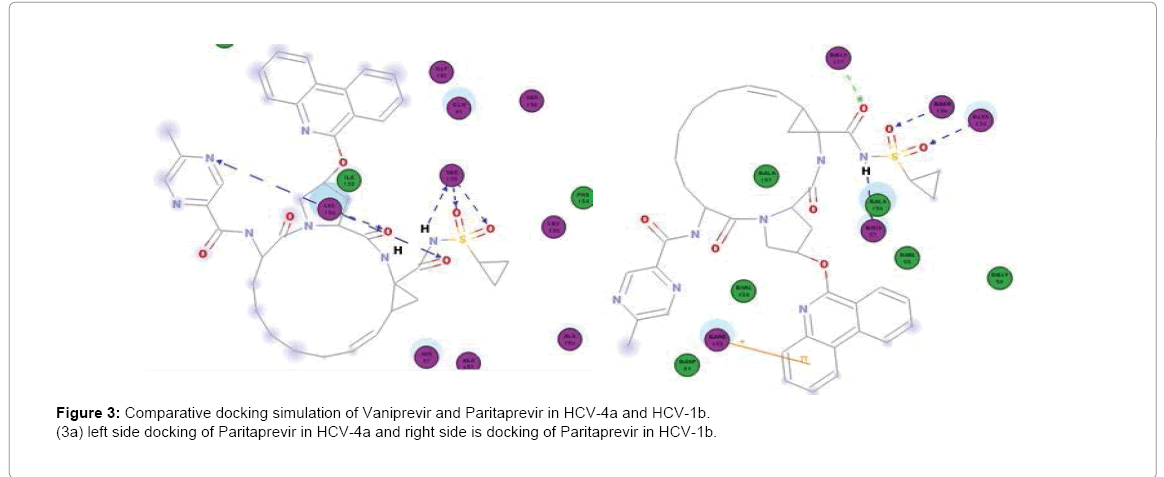
Figure 3: Comparative docking simulation of Vaniprevir and Paritaprevir in HCV-4a and HCV-1b.
(3a) left side docking of Paritaprevir in HCV-4a and right side is docking of Paritaprevir in HCV-1b.
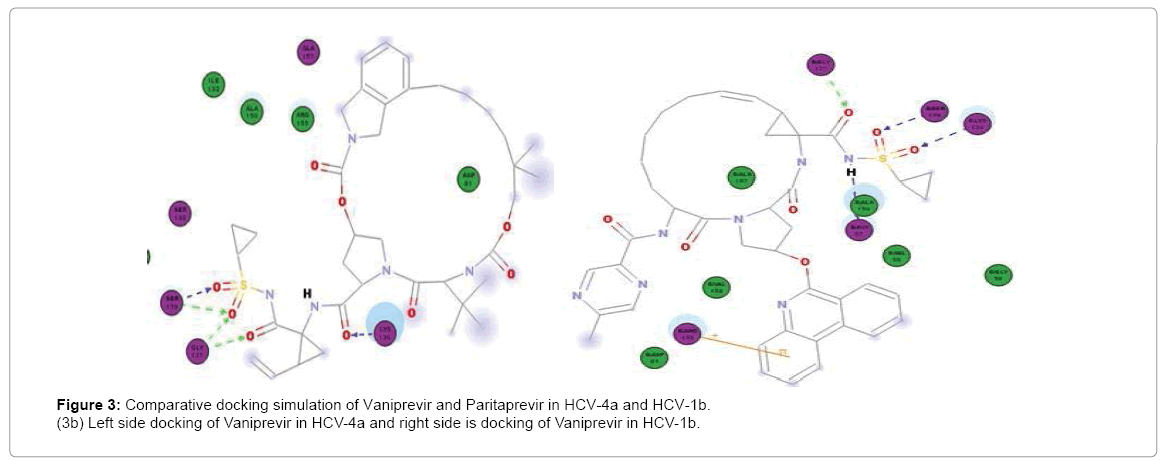
Figure 3: Comparative docking simulation of Vaniprevir and Paritaprevir in HCV-4a and HCV-1b.
(3b) Left side docking of Vaniprevir in HCV-4a and right side is docking of Vaniprevir in HCV-1b.
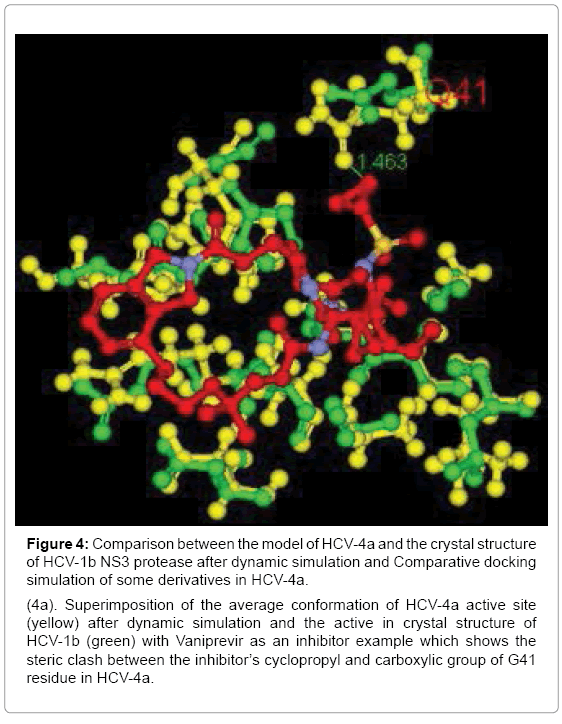
Figure 4: Comparison between the model of HCV-4a and the crystal structure of HCV-1b NS3 protease after dynamic simulation and Comparative docking simulation of some derivatives in HCV-4a.
(4a). Superimposition of the average conformation of HCV-4a active site (yellow) after dynamic simulation and the active in crystal structure of HCV-1b (green) with Vaniprevir as an inhibitor example which shows the steric clash between the inhibitor’s cyclopropyl and carboxylic group of G41 residue in HCV-4a.
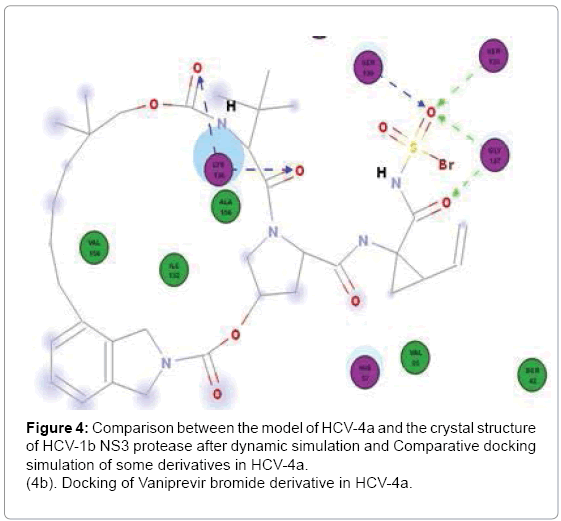
Figure 4: Comparison between the model of HCV-4a and the crystal structure of HCV-1b NS3 protease after dynamic simulation and Comparative docking simulation of some derivatives in HCV-4a.
(4b). Docking of Vaniprevir bromide derivative in HCV-4a.
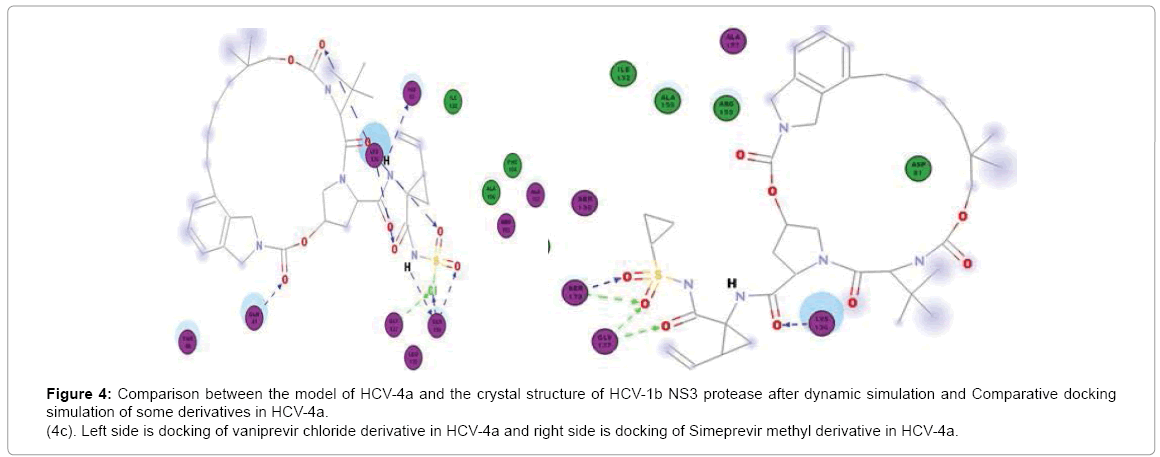
Figure 4c: Comparison between the model of HCV-4a and the crystal structure of HCV-1b NS3 protease after dynamic simulation and Comparative docking simulation of some derivatives in HCV-4a.
(4c). Left side is docking of vaniprevir chloride derivative in HCV-4a and right side is docking of Simeprevir methyl derivative in HCV-4a.
Table 1 provides the interactions between the modified inhibitors and the residues in the active site of NS3 of GT4a.
| Inhibitor | Cyclopropyl replaced by | Residues involved in H-bond interactions | Residues involved in pi-interactions | LibScore |
| Chloride | K136(2), S139(2) | H57 | 134.1 | |
| Bromide | K136(2), G137(2), S139(2) | H57 | 138 | |
| methyl | H57,R123,K136(2), G137, S139 | R155 | 128.3 | |
| Hydroxyl methyl | H57, K136(3), G137(1), S139(2) | 143.6 | ||
| Vaniprevir | Fluoride | H57, K136(2), S139(2) | K136 | 121.3 |
| Chloride | Q41, H57, K136(3), G137, S139(3) | 152.9 | ||
| Bromide | K136(2), G137(2), S138, S139 | 115.8 | ||
| methyl | Q41, K136,G137(2), S139 | H57 | 143.6 | |
| Hydroxyl methyl | K136(2), G137, S139(3) | H57, R155 | 170.5 | |
| Paritaprevir | Fluoride | K136(2), G137, S139(2) | H57 | 138.3 |
| Chloride | Q41, K136, G137(2), S139(3) | 142.9 | ||
| Bromide | K136, G137(2), S138, S139 | 117.6 | ||
| methyl | H57, K136, G137(2), S139 | 140.3 | ||
| Hydroxyl methyl | Q41, H57, K136, G137(2), S139 | 145.7 |
Table 1: Provides the interactions between the modified inhibitors and the residues in the active site of NS3 of GT4a. Residues involved in H-bond and Pi-interactions with inhibitors are indicated and number of H-bonds for residues forming more than one H-bond is shown within the brackets.
The differences in the binding mode of the three drugs to both the genotypes are due to significant variability in the conformation of some amino acid residues in the active site. We performed dynamic simulation of the 3D model to ensure that the result of docking is due to steric clash with Q41 and distance increase between H57 and S139 in NS3 of GT4a.
Dynamic simulation
However the NS3A protease of GT4a was highly dynamic but the conformation change in Q41 residue of the average conformation of the 20 copies relative to the H57 and S139 was very little and maintained the steric clash with cyclopropyl of the inhibitors with slight distance increase (Figure 4a) and this indicates that the conformational change in the active site of HCV NS3 of GT4a from GT1b is responsible of different drug activity in both the genotypes.
There are conformational differences between residues in the active site of NS3 HCV protease in GT1b and GT4a. These differences enable the inhibitors to be active in GT1b but hinder their activity in GT4a and so that Structure Based Drug Design could be used for developing new inhibitors that act specifically for GT4a.