Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2012) Volume 5, Issue 3
Gene Set Enrichment Analysis (GSEA) utilizes the gene expression profiles of functionally related gene sets in Gene Ontology (GO) categories or prior defined biological classes to assess the significance of gene sets associated with clinical outcomes or phenotypes and are the most widely used method for gene analysis. However, little attention has been given from a classification prospect. In this paper, we identify the differential gene sets, which are strongly associated with phenotypic class distinction ability, using gene expression data together with prior biological knowledge. We propose two non-parametric methods to identify differential gene sets using the area under the receiver operating characteristic (ROC) curve (AUC) of linear risk scores of gene sets, which are obtained through a parsimonious threshold-independent gene selection method within gene sets. The AUC-based statistics and the AUC values obtained from cross-validation of the linear risk scores are calculated, and used as indexes to identify differential gene sets. The discrimination abilities of gene sets are summarized and gene sets that possess discrimination ability are selected via a prescribed AUC statistic threshold or a predefined cross-validation AUC threshold. Moreover, we further distinguish the impacts of individual gene sets in terms of discrimination ability based on the absolute values of linear combination coefficients. The proposed methods allow investigators to identify enriched gene sets with high discrimination ability and discover the contributions of genes within gene set via the corresponding linear combination coefficients. Both numerical studies using synthesized data and a series of gene expression data sets are conducted to evaluate the performance of the proposed methods, and the results are compared to the random forests classification method and other hypotheses testing based approaches. The results show that our proposed methods are reliable and satisfactory in detecting enrichment and can provide an insightful alternative to gene set testing. The R script and supplementary information are available at http://idv.sinica.edu.tw/ ycchang/software.html.
Keywords: Gene set enrichment analysis; The receiver operating characteristic (roc) curve; The area under the roc curve (Auc); Discrimination ability; Cross-validation; Linear combination coefficients; Random forests classification method
Biological phenomena often occur through the interactions of multiple genes via signalling pathways, networks, or other functional relationships. Based on that information, a set of genes with related functions is grouped together and referred to as a "gene set"; among them, if the expression level of such a gene set is significantly associates with the clinical outcomes/phenotypes, then we say that this gene set is "differentially expressed". Thousands of genes that share common functional annotation are organized into groups (possibly overlapping), and the information for grouping genes as gene sets can be obtained through publicly available annotation databases such as the Kyoto Encyclopaedia of Genes and Genomes (KEGG), Gene Ontology (GO), and GenMAPP.
Many statistical approaches, such as gene set analysis (GSA) methods, are used to determine whether such functionally related gene sets express differentially (enrichment and/or deletion) in variations of phenotypes, and a common approach of GSA methods is first to identify a list of genes that express differently among two groups of samples using a statistical test, and this list of differentially expressed genes is then examined with biologically pre-defined gene sets to determine whether any set in the list is over-represented compared with the whole list of gene sets [1-3]. These types of approaches do not consider the correlation structure and the order of genes in a gene set. Therefore, Mootha et al. [4] and Subramanian et al. [5], in order to improve on GSA, proposed the Gene Set Enrichment Analysis (GSEA), in which they consider the distributions of entire genes in a gene set, rather than a subset from the list of differential expression genes, and use some statistic to assess the significance of predefined gene sets.
Following the idea of GSEA, many statistical methods have been proposed, such as the global test [6], the two-sample t-test like approach [7], the ANCOVA test [8], the Hotelling's T2 test [9], the MaxMean approach [10], the SAM-GS test [11], the global statistics approach [12], the Random-sets method [13], the Logistic Regression (LRpath) approach [14], and the MANOVA test [15] amongst others. These approaches rely on different statistical assumptions and data structures, which then usually lead to different results even when they are applied to the same data set. For the underlying assumptions and a comprehensive review of these methodologies can be found in, for example, Goeman and Buhlmann [16] and Nam and Kim [17]. From their papers, we note that none of the methods have addressed the feasibility of discriminating different phenotypes via a priori defined gene sets.
On the other hand, there various machine learning type algorithms have been developed from a classification prospective. For example, Lin et al. [18] demonstrated that the classification accuracy and robustness of classification in analyzing microarray data can be improved by considering the existing biological annotations, Pang et al. [19,20] used the random forests classification and regression, Wei and Li [21] applied a boosting-based method for nonparametric pathways-based regression (NPR) analysis, and Tai and Pan [22,23] proposed a group penalization method that incorporates biological information to build a penalized classifier to improve prediction accuracy. In particular, Lottaz and Spang [24] provided a biologically focused classifier, say StAM, based on the GO hierarchical structure, and only the genes annotated in the leaf nodes of the GO tree can be used as predictors, and other genes (relevant, but not annotated yet), cannot be used in their method. Since the biological information of gene sets may come from different databases, such as KEGG or BioCarta, and not limited to the GO annotation only, therefore, when the GO terms of genes in gene sets are unavailable, this type of method may result in losing information about the predictive model. Although, NPR aims to improve on the predictive accuracy via incorporation of biological knowledge, there is no selection criterion with respect to identification of differential gene sets.
In this paper, we propose a GSA method that can not only to identify gene sets that have only a subset of genes with expression profiles, which are strongly associated with the class distinction, but also to increase the discrimination ability such that subjects with different phenotypes or clinical outcomes are appropriately classified by integrating the selected gene sets together. During the analysis, the proposed method treats each gene set as a whole, while retaining ability to interpret impacts of individual genes on a prediction model. Here we assume the data set contains the a priori biological information of gene sets. For data sets without gene sets information, the proposed method can still be applied in the same manner, if an appropriate clustering algorithm can be applied to obtain gene clusters first. However, the focus of this type of analysis is usually different from that of analyzing data with intrinsic gene set information. The details are given in the web-supplement.
It is well-known that the AUC, as a summary index, shares the threshold independent characteristic of ROC curves. Hence, in this paper, we propose this AUC-based method for identifying differential gene sets and study the detailed procedure of selecting gene sets with discrimination ability. In addition, AUC-based statistics are proposed and can be used to assess and rank the significance of gene sets. The remainder of this paper is organized as follows: the background of proposed methods and the two new selection procedures are given in Section 2. In Section 3, the performances of our proposed approach are compared to the random forests-based pathway analysis approach (pathwayRF) [19] based on the synthesized data sets and a series of real expression data. In addition, our method is also compared to other hypotheses testing based approaches such as global ANCOVA gene set testing [8] and the rotation gene set testing [16]. A discussion of the proposed approach is presented in Section 4. For other properties of AUC and its applications, please refer to Metz et al. [25], Su and Liu [26], Zhou et al. [27], Pepe [28], Liu et al. [29], Ma and Huang [30] and Wang et al. [31], and the references therein.
Suppose that each subject has p observed continuous-valued outputs of genes in a binary classification problem, say diseased and normal classes. Assume further that these p genes belong to one of K predefined gene sets based on, for example, GO category. The proposed method consists of the following three steps:
(i) constructing a classifier with continuous decision output based on genes within each gene set;
(ii) evaluating the discrimination ability of all gene sets to detect diseased and normal groups based on the classifiers obtained in (i);
(iii) identifying the sets with high discrimination ability through construction of a classifier ensemble using the classifiers obtained in (i).
After step (i), each gene set is represented by a function of the classifier constructed using the genes of this gene set. That is, we treat each gene set as a unit simply by taking the output values of the function value of the corresponding classifier as a new feature, and subjects are represented using these new features. Therefore, a linear ensemble of these new features that maximizes the AUC is constructed, which has the best classification performance in terms of AUC. In step (iii), we construct a linear ensemble of classifiers based on these new features. We then identify gene sets according to their contributions to this final ensemble, which can usually be indexed using the corresponding coefficients of the linear combination. Furthermore, another crossvalidation schemes are also recommended to assess the identification stability of differential gene sets.
Please note that if we are only interested in gene sets identification, then the only requirement of the base-classifiers used in step (i) is being able to provide continuous classification function values, otherwise there is no limitation on the usages of classification methods in the proposed method. Thus, many classification methods, such as linear discrimination, logistic regression, support vector machines and others, are applicable in this case. Since different classification methods will usually select/utilize genes in different ways such that the maximum classification performance can be achieved, then it makes the assessment of the impact of individual genes very difficult if different classification methods are used within different gene sets. In this paper, in order to fully take advantage of the threshold-independence of the ROC curve and retain the interpretation ability of each gene, we adopt the best linear combination of genes that maximizes AUC within each gene set for constructing the base classifiers in (i). Thus, our method not only identifies the differential gene sets, but also provides the measure of importance of genes within each gene set. Moreover, in the web supplement we also illustrate how to apply our method when there is no intrinsic grouping information available, in which the K-means and a hierarchical clustering methods are applied to obtain gene sets.
The area under the ROC curve
Let W be a continuous-valued random variable, and suppose that for a pre-specified threshold c, a subject is classified as diseased (positive) if W>c, or normal (negative) otherwise. Then the ROC curve for such a simple classifier W is 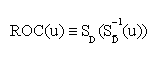 , where
, where 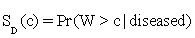 and
and 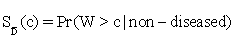 are functions of the true positive and false positive probabilities, respectively. The
are functions of the true positive and false positive probabilities, respectively. The 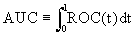 is defined as an integration of true positive rate over the whole range of the false positive rate. Let function ψ(u)=1 if u>0; =0.5 if u=0 and =0 if u<0 and {WD,1,...,WD,n,WN,1,...,WN,m} be observations of n diseased and m normal subjects. Then it is wellknown that the AUC can be estimated using the Wilcoxon-Mann- Whitney U-statistic
is defined as an integration of true positive rate over the whole range of the false positive rate. Let function ψ(u)=1 if u>0; =0.5 if u=0 and =0 if u<0 and {WD,1,...,WD,n,WN,1,...,WN,m} be observations of n diseased and m normal subjects. Then it is wellknown that the AUC can be estimated using the Wilcoxon-Mann- Whitney U-statistic
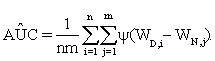 (1)
(1)
It is shown in Kowalski and Tu [32] that
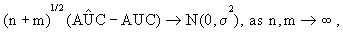 (2)
(2)
where 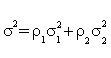 , ρ1 and ρ2 are the limits of (n + m) / n and (n + m) / m , respectively, with
, ρ1 and ρ2 are the limits of (n + m) / n and (n + m) / m , respectively, with
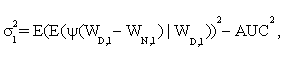
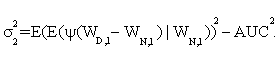
In addition, it is shown that asymptotic standard deviation σ can be estimated by
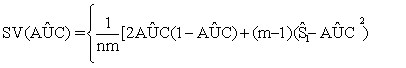
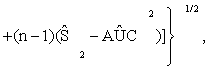 (3)
(3)
where
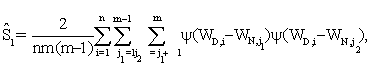
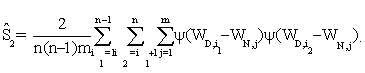
The detailed arguments are given in the web supplement
Linear combination of genes within a gene set
Suppose that each gene set has pk genes, and Xk's and Yk's are pk -dimensional random vectors of gene expressions data of the k-th gene set from two (normal and diseased) groups, k=1,...,K. As mentioned in (i), for each k, we first construct a linear classifier to discriminate between these two different groups by using the pk genes within k-th gene set. Let lk be a vector of constants with length pk, then the linear combinations l'kXk andl'kYk are called risk scores. There is usually no distribution information available for Xk and Yk, so the best lk,for each k, that achieves the maximum AUC among all possible pk -dimensional vectors must be calculated through observations. Let {Xkj,Yki: j=1,...,m;i=1,...,n}be observed expression values of genes in the k-th gene set of the two groups. From (1), the empirical AUC estimate can be rewritten as
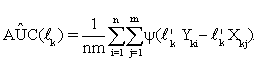 (4)
(4)
Since step function ψ(·) is not continuously differentiable, to compute such a linear combination coefficient by directly maximizing AÛC(lk) is difficult. Thus, we follow Ma and Huang [30] and Wang et al. [31], to use a sigmoid function S(t)=1/(1+exp(-t)) to approximate ψ(·). Consequently, the smoothed AUC estimate is defined as,
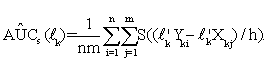 (5)
(5)
It has been shown in Wang et al. [31] that for sufficiently small h, S((y-x)/h)≈ψ((y-x) and AÛCs(lk) is a strongly consistent estimator of AUC. So, we obtain a vector ˆ lk of the ''optimal'' linear combination coefficients for the k-th gene set through maximizing the smoothed AUC estimate (5) with respect to lk; that is,
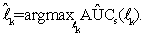
Since AUC is scale invariant, AÛC(lk) has the same value as AÛC(clk) for any positive constant c. Hence, an anchor gene should be determined before finding the solution that maximizes AÛCs(lk) such that lk is identifiable. However, when the number of genes in one set is more than the total sample size, e.g. pk>>(n+m), obtaining vector 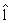 k by direct maximization of (5) could lead to over-fitting. Therefore, we use the PTIFS method proposed in Wang et al. [31] to find the ``optimal'' ˆ lk for each k. We describe a summary of the PTIFS algorithm based on the k-th gene set with pk genes as following, details seen in Wang et al. [31], where we assume that gene 1 is the anchor gene and denote sets of active genes and inactive genes at the iteration procedure by Ak and Akc , respectively.
k by direct maximization of (5) could lead to over-fitting. Therefore, we use the PTIFS method proposed in Wang et al. [31] to find the ``optimal'' ˆ lk for each k. We describe a summary of the PTIFS algorithm based on the k-th gene set with pk genes as following, details seen in Wang et al. [31], where we assume that gene 1 is the anchor gene and denote sets of active genes and inactive genes at the iteration procedure by Ak and Akc , respectively.
Algorithm
(i) Find the anchor gene which is Ak= {1} as assumed and set Gk =φ, the empty set.
(ii) For the current active set Ak, calculate the corresponding coefficients,kA , of the linear classifier by the criterion function (6). For each j∈ Akc , compute Rj=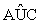 (lk ) , the empirical AUC for the j-th gene based on all the subjects of the two groups; and compute its empirical AUC, Rj(0) , based on subjects that are misclassified by A kA , where lk=βkj is a vector of length pk with the j-th component being 1 and the others being 0.
(lk ) , the empirical AUC for the j-th gene based on all the subjects of the two groups; and compute its empirical AUC, Rj(0) , based on subjects that are misclassified by A kA , where lk=βkj is a vector of length pk with the j-th component being 1 and the others being 0.
(iii) If 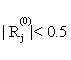 for all
for all 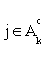 or
or 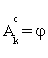 , then stop. Otherwise choose
, then stop. Otherwise choose 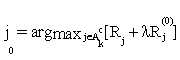 ,and update the active set Ak by adding the feature j0 and excluding it from the inactive set
,and update the active set Ak by adding the feature j0 and excluding it from the inactive set 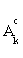 , where λ>0 is a pre-specified constant weighting on the misclassified subjects.
, where λ>0 is a pre-specified constant weighting on the misclassified subjects.
(iv) Update 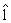 kA l by using objective function (6) with respect to the updated active set Ak in step
kA l by using objective function (6) with respect to the updated active set Ak in step
(iii). Remove the set 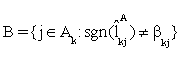 from Ak and add it to inactive set If j0∈B , then exclude j0 from and add j0 to Gk, otherwise add the elements of Gk to and let Gk=φ.
from Ak and add it to inactive set If j0∈B , then exclude j0 from and add j0 to Gk, otherwise add the elements of Gk to and let Gk=φ.
(v) Repeat (ii)-(iv) until AÛC(kA)≥τ , where 0.5 ≤τ < 1 .
In many biological studies, such as treatment/drug developments, the assessment of individual genes is highly valued. When a nonlinear classifier is used, it is usually difficult to distinguish the impacts of individual genes due to the complicated classification function, which makes designing the follow-up studies on individual genes very difficult. That is another reason, in addition to the technical ones, why the linear classifier is preferred in the proposed method. It is clear that in this case, the impact of both gene sets and individual genes can be identified and follow-up experiments can be easily planned based on the findings. PTIFS is adopted when the follow-up biological confirmation is of concern. Because of its parsimonious property, PTIFS may also benefit biological researchers designing the necessary and expensive experiments. AUC is chosen as an indicator of performance here due to its threshold independent property. Moreover, the AUC can be calculated easily such that a test statistic can be founded on that. However, the AUC can be replaced by other performance measures without any difficulty as long as a method of calculating coefficients of a linear combination vector under such a measure is available.
Assessment of gene set significance
According to the methods used to maximize AUC, we use 3 measures to assess the significance of a gene set, which are AUC statistic of gene set, cross-validation AUC of gene set, and coefficients of linear combination of gene sets. Once step (i) is completed for all K gene sets, we have k for each k =1,...,K. The AÛCk= AÛCk is then calculated using (4). For each k, define statistic zk=AÛCk–0.5, which will be used as an index for identifying differential gene sets. It is known that if gene set k has no discrimination ability to distinguish the diseased subjects from the normal ones, then its corresponding ROC curve is usually close to the diagonal line of the unit square and the AUC approximately equals 0.5. The larger the zk, the better the classification performance of gene set k. Thus, we can select the top-ranking gene sets according to zk until a prescribed threshold is reached. For a given linear combination lk of genes in gene set k, hence, from (2), AÛC(lk) is asymptotically normally distributed. However, AÛCk is a minimizer of AÛC(lk) with respect to lk and the asymptotic distribution of AÛCk is difficult to be computed. Therefore, if a p value of gene set must be needed, then a permutation-based approach can be employed to calculate the empirical p-value for each gene set by randomly drawing gene sets with the same size as gene set k.
In practice, the size of genes in a gene set may be much larger than the number of subjects available for analysis. Thus, to prevent overfitting so that the corresponding AÛC will not be over-optimistically large, the cross-validation method should be used. Following the usual cross-validation scheme and applying the constructed classifier to the testing sample, the predicting AUC, say AUCcv,k, is calculated for each k. Since the larger the AUCcv,k, the higher the discriminant potential of the k-th gene set, we can now select gene sets according to AUCcv,k as in the zk cases.
To construct the final classification ensemble in (iii), we first represent all individual subjects by their corresponding function values after applying the classifiers obtained from individual gene sets to all subjects. So, those classification scores of each subject are treated as a set of new variables. The final ensemble is an integration of all gene sets using PTIFS, which provides us a linear combination of these new variables such that the final AUC is maximized. Consequently, the gene sets are ranked according to absolute values of linear combination coefficients, and the top-ranked ones are selected. The interpretation ability of individual genes is retained due to a linear combination is used in each stage. Note that once we represent subjects by function values of the gene set based classifiers, the dimensionality of these new variables is reduced to K -- the total number of gene sets considered, which is usually much smaller than the size of genes considered.
Simulation study
We evaluate the performance of the proposed method using an extensive simulation study. For the evaluating purposes, N= 10000 genes are generated from a multivariate normal (MVN) distribution with a mean vector μ and a diagonal variance-covariance matrix Σ for both diseased and normal groups, and only a fraction out of N genes, say γ ∈ (0,1), is differentially expressed with a mean difference δ. The sets S0 and S1 respectively contain the non-differential and differential genes. Thus, there are 10000γ genes in set S1 and 10000(1-γ) genes in set S0. The diagonal elements of Σ are equal to 1. Genes in set S1 are correlated with a correlation coefficient ρ |i-j| between gene i and gene j. In this study, ρ is set to 0 (i.e., independent model) and 0.5. The
informative gene set is composed of 100 genes, where 100γ genes are randomly selected from set S1 and the other 100(1-γ) genes are from set S0. This procedure is then repeated 10 times. Additionally, we generate 10 non-informative gene sets in which 100 genes are randomly selected from set S0. In summary, we generate 20 gene sets in each (diseased and normal) groups, and there were 100 genes in each gene set. In the 20 simulated gene sets, only the first 10 sets has discriminant ability, and only the first 100γ genes in these gene sets are differently expressed. In our simulation studies, γ is set to be 0.05 and 0.10, as well as δ is 0.0, 1.0, and 1.5. We note that, when δ=0, the Type I error rates of competing methods is investigated. The training sample sizes are n=m=50 and testing sample sizes are n=m=20 for both diseased and normal groups. All simulations are repeated 200 times.
We investigate, from a classification prospect, the performance in identifying differential gene sets of the proposed method, and compare with those in pathway analysis using random forests classification (pathwayRF) [19] the least square support vector machine (ls-svm), global gene set testing (Global ANCOVA) [8] and the rotation gene set testing (Roast) [16]. We use the out-of-bag (OOB) error estimate, cross validation error rate, F-test statistic and p value to assess the discriminant abilities of gene sets for methods pathwayRF, ls-svm, Global ANCOVA and Roast, respectively. The OOB estimate is based on recording the votes of classification on the test set left out of the bootstrap sample. For each gene set, based on training data sets, we calculate the fivefold cross-validation AUC, AUCcv using parsimonious threshold independent feature selection (PTIFS), OOB as that in the pathwayRF method, cross validation error rate for the ls-svm, F -test statistic for the Global ANCOVA method and p value in the Roast method. The PTIFS, proposed in Wang et al. [31], is a LARS-type algorithm that helps to find the linear combination of markers that maximizes AUC. Note that AUC is not available for pathwayRF due to the complicated voting scheme of the random forests process. Since Global ANCOVA and Roast are based on views of regression, they can not be used to make prediction for two-group classification data. We then compare the accuracies of detecting gene sets of these five approaches, where the accuracy is defined as the percentage of correctly identified gene sets. In Figures 1 and 2 with ρ =0.0 and 0.5, the upper three panels and the first two panels in the lower respectively present mean accuracy of AUCcv, pathwayRF, ls-svm, Global ANCOVA and Roast for γ= 0.05 or 0.10 and δ= 1.0 or 1.5. We can see that the accuracy is greater than 0.95 for AUCcv with cut-point in interval (0.60, 0.75), for pathwayRF with cut-point in interval (0.30, 0.40), for Global ANCOVA with cutpoint in (1.0, 2.0) and for Roast with cut-point in (0.01, 0.06) except the parameter combination (γ =0.05, δ =1.0). Method ls-svm has the least accuracy smaller than 0.52 which suggests that ls-svm is not suitable to identify differentially expressed gene set. Hence, the proposed AUCcv is a promising and reliable measure to identify differential gene sets. At the same γ, AUCcv with larger δ =1.5 has higher accuracy than those of the cases with smaller δ( =1.0). For the same δ, AUCcv with larger γ(= 0.10) has greater accuracy than those with smaller γ(= 0.05). This is because AUCcv is sensitive to the discrimination ability of gene set. Similarly, the larger δ for given γ or the larger γ for given δ, the greater accuracy; and pathwayRF, Global ANCOVA and Roast also perform well in these cases. In addition, the k AÛC for the k-th gene set is computed and the statistic zk = AÛCk -0.5 is calculated. The third panel in the lower of Figures 1 and 2 presents the mean accuracy values of based on zk with γ= 0.05 or 0.10, and δ= 1.0 or 1.5. It is found that, except situation of γ= 0.05 and δ= 1.0, all zk have accuracies greater than 0.90 for any cut-point value in the interval (0.43, 0.45), which indicates that the zk performs well for identifying differential gene sets. Table 1 presents the average fitting and prediction error rates of the top-ten gene sets selected by AUCcv, pathwayRF and ls-svm. The results again confirms that our proposed AUCcv provides competitive classification results, in terms of the classification errors, to that of pathwayRF, and much better than that of ls-svm. Table 2 shows the empirical type I errors of three methods, Global ANCOVA, Roast and AUC statistic zk, using the nominal level of 0.05. Note that the p value of zk is computed by using permutation method. As expected, the type I error rates of all three methods are reasonably close to or below the nominal level in all settings.

Figure 1: Results of simulation study with correlation coefficient ρ=0.0. Performance comparisons of PTIFS, pathwayRF, ls-svm, Global ANCOVA and Roast methods Accuracy of identifying gene set for cross-validation AUC, p-values of AUC statistic, pathway RF method, ls-svm method, global F statistics and p-values of roast method.

Figure 2: Results of simulation study with correlation coefficient ρ=0.5. Performance comparisons of PTIFS, pathwayRF, ls-svm, Global ANCOVA and Roast methods Accuracy of identifying gene set for cross-validation AUC, p-values of AUC statistic, pathway RF method, ls-svm method, global F statistics and p-values of roast method.
| Fitting | Predicting | ||||||||
| ρ | Method | γ | δ | Error rate | AUC | Error rate | AUC | ||
| 0.0 | AUC | 0.05 | 1.0 | 0.106 | 0.952 | 0.188 | 0.899 | ||
| 0.05 | 1.5 | 0.043 | 0.991 | 0.082 | 0.976 | ||||
| 0.10 | 1.0 | 0.052 | 0.987 | 0.128 | 0.946 | ||||
| 0.10 | 1.5 | 0.036 | 0.994 | 0.093 | 0.971 | ||||
| pathwayRF | 0.05 | 1.0 | 0.217 | (-) | 0.194 | (-) | |||
| 0.05 | 1.5 | 0.085 | (-) | 0.076 | (-) | ||||
| 0.10 | 1.0 | 0.117 | (-) | 0.103 | (-) | ||||
| 0.10 | 1.5 | 0.025 | (-) | 0.021 | (-) | ||||
| ls-svm | 0.05 | 1.0 | 0 | 1 | 0.456 | 0.559 | |||
| 0.05 | 1.5 | 0 | 1 | 0.428 | 0.598 | ||||
| 0.10 | 1.0 | 0 | 1 | 0.434 | 0.592 | ||||
| 0.10 | 1.5 | 0 | 1 | 0.399 | 0.643 | ||||
| 0.5 | AUC | 0.05 | 1.0 | 0.131 | 0.930 | 0.233 | 0.852 | ||
| 0.05 | 1.5 | 0.058 | 0.983 | 0.112 | 0.958 | ||||
| 0.10 | 1.0 | 0.070 | 0.976 | 0.170 | 0.910 | ||||
| 0.10 | 1.5 | 0.041 | 0.992 | 0.102 | 0.964 | ||||
| pathwayRF | 0.05 | 1.0 | 0.245 | (-) | 0.222 | (-) | |||
| 0.05 | 1.5 | 0.116 | (-) | 0.103 | (-) | ||||
| 0.10 | 1.0 | 0.144 | (-) | 0.125 | (-) | ||||
| 0.10 | 1.5 | 0.044 | (-) | 0.038 | (-) | ||||
| ls-svm | 0.05 | 1.0 | 0 | 1 | 0.467 | 0.552 | |||
| 0.05 | 1.5 | 0 | 1 | 0.442 | 0.586 | ||||
| 0.10 | 1.0 | 0 | 1 | 0.445 | 0.577 | ||||
| 0.10 | 1.5 | 0 | 1 | 0.416 | 0.620 | ||||
*Here we use error rate to denote the misclassification rate.
Table 1: Simulation results. Average fitting and predicting error rates of top-10 gene sets selected by AUCcv, pathwayRF and ls-svm.
| Global | ||||
| ρ | γ | ANCOVA | Roast | AUC |
| 0.0 | 0.05 | 0.055 | 0.055 | 0.040 |
| 0.10 | 0.040 | 0.040 | 0.058 | |
| 0.5 | 0.05 | 0.025 | 0.040 | 0.058 |
| 0.10 | 0.055 | 0.045 | 0.050 |
Table 2: Simulation results. Empirical type I errors of three methods, Global ANCOVA, Roast and AUC statistic zk.
When ρ = 0.0 and 0.5, as an example, Figures 3 and 4 respectively describe the average values of within-gene-set coefficients for the first gene set (gene set 1) obtained using PTIFS with γ= 0.05 and 0.10 and δ= 1.0 and 1.5. In these figures, the vertical lines represent the positive (up) and negative (down) values of the coefficient, respectively, and the length of lines indicates the magnitude of the absolution value of the coefficient within each gene set. When γ= 0.05 (0.10), the magnitudes of coefficients of the first 5 (10) genes are the largest since only the first 5 (10) genes have discrimination ability. For the same δ, the magnitude of the coefficient of genes selected at γ= 0.05 is bigger than that at γ= 0.10. This is because when the fraction γ is large, the number of differentially expressed genes selected increases and the number of candidate genes also increases. So, the selection frequencies become smaller than those of the case with small γ, due to the parsimonious property of PTIFS. By applying PTIFS to the newly extracted variables using the base-classifiers of individual gene sets, we are able to select the differentially expressed gene sets. Table 3 lists the number of gene sets selected (selnum) based on PTIFS, AUC and the misclassification rate of the linear classifier using both training and testing data sets. It shows that our method performs well in terms of classification error rate.

Figure 3: Within-gene-set coefficients for simulation study with correlation coefficient ρ=0.0. The average values of within-set coefficients obtained by the PTIFS algorithm for gene set 1.

Figure 4: Within-gene-set coefficients for simulation study with correlation coefficient ρ=0.5. The average values of within-set coefficients obtained by the PTIFS algorithm for gene set 1.
| Fitting | Predicting | |||||||||
| ρ | γ | δ | selnum | AUC | Error | AUC | Error | |||
| rate | rate | |||||||||
| 0.00 | 0.05 | 1.0 | 1.071 | 0.992 | 0.041 | 0.843 | 0.226 | |||
| (0.258) | (0.003) | (0.014) | (0.162) | (0.140) | ||||||
| 0.05 | 1.5 | 1 | 0.996 | 0.027 | 0.978 | 0.077 | ||||
| (0) | (0.002) | (0.011) | (0.022) | (0.045) | ||||||
| 0.10 | 1.0 | 1 | 0.995 | 0.033 | 0.951 | 0.122 | ||||
| (0) | (0.002) | (0.011) | (0.035) | (0.055) | ||||||
| 0.10 | 1.5 | 1 | 0.998 | 0.019 | 0.975 | 0.088 | ||||
| (0) | (0.001) | (0.011) | (0.021) | (0.046) | ||||||
| 0.50 | 0.05 | 1.0 | 1.162 | 0.991 | 0.042 | 0.773 | 0.289 | |||
| (0.418) | (0.003) | (0.014) | (0.168) | (0.14) | ||||||
| 0.05 | 1.5 | 1 | 0.995 | 0.033 | 0.965 | 0.102 | ||||
| (0) | (0.002) | (0.011) | (0.027) | (0.048) | ||||||
| 0.10 | 1.0 | 1.005 | 0.993 | 0.040 | 0.919 | 0.160 | ||||
| (0.069) | (0.002) | (0.012) | (0.058) | (0.070) | ||||||
| 0.10 | 1.5 | 1 | 0.997 | 0.024 | 0.967 | 0.095 | ||||
| (0) | (0.001) | (0.011) | (0.027) | (0.049) | ||||||
Table 3: Simulation results. Classification results using the PTIFS gene set selection method. The numbers in parentheses show the standard deviations.
Real examples
We apply our method to data sets obtained from six microarray gene expression studies with intrinsic gene sets, which are Gender, p53, Diabetes, Leukemia, lung cancer from the Boston study and lung cancer from the Michigan study. All these data sets are frequently used in gene set analysis [4,5,11,15]. In each study, the gene sets are clustered into several sets based on two catalogs: (C1) chromosomes and cytogenetic catalog, and (C2) functional catalog. The gender data set consists of 15 males and 17 females, and each sample has 15,056 mRNA expression profiles. The p53 data set is from a study identifying targets of the transcription factor p53 from 10,100 gene expressions with 17 normal and 33 mutation samples. In the diabetes study, the DNA microarrays are used to profile expressions of 15,056 genes from 34 skeletal muscle biopsy samples (17 normals and 17 patients). The leukemia data set includes 10,056 expression profiles from 24 acute lymphoblastic leukemia (ALL) patients and 24 acute myeloid leukemia (AML) patients. The lung cancer data set is obtained from studies conducted by the Boston and Michigan groups. The Boston group studied 5,217 gene expression levels from 31 dead and 31 live subjects, and the Michigan group studied lung tumors by comparing 5,217 gene expression profiles derived from 24 dead subjects with those of 62 live subjects.
We apply both the AUCcv and pathwayRF methods to the seven data sets for identification of differential gene sets: Gender (C1), Gender (C2), p53 (C2), diabetes (C2), acute leukemia (C1), and lung cancer (C2) from the Boston study, and lung cancer (C2) from the Michigan study. The number of gene sets are 212, 318, 308, 318, 182, 258 and 258, respectively. For each gene set, we use the PTIFS algorithm to obtain an optimal linear combination coefficients of genes. We obtain a 5-fold cross-validation estimate of AUC, AUCcv, and apply pathwayRF with OOB values. We then rank all gene sets via AUCcv and OOB. Tables 4 and 5 show the top-ten ranking gene sets based on AUCcv and the smallest OOB for these seven gene expression data sets, respectively.
| Rank | Gender C1 | Gender C2 | P53 | Diabetes |
| 1 | chrY | gnf female genes | calcineurinpathway | p53 down |
| (0.987, 0.031) | (0.995, 0) | (0.895, 0.12) | (0.812, 0.324) | |
| 2 | chrYp11 | testis genes from | human mitodb 6 | map00251 gluta- |
| (0.987, 0.031) | xhx and netaffx | 2002(0.893, 0.16) | mate metabolism | |
| (0.987, 0.062) | (0.801, 0.353) | |||
| 3 | chrYq11 | xinact merged | mitochondr(0.887, 0.16) | vippathway |
| (0.987, 0.062) | (0.92, 0.156) | (0.787, 0.353) | ||
| 4 | chrXp22 | prolif genes | bcl2family and reg | xinact merged |
| (0.908, 0.125) | (0.916, 0.156) | network(0.871, 0.12) | (0.775, 0.324) | |
| 5 | chrX | st dictyostelium disco- | ceramidepathway | p53 signalling |
| (0.896, 0.156) | ideum camp chemotaxis | (0.864, 0.1) | (0.765, 0.324) | |
| pathway(0.916, 0.156) | ||||
| 6 | chrXp11 | cell proliferation | badpathway(0.861, 0.1) | mcalpainpathway |
| (0.837, 0.312) | (0.916, 0.094) | (0.763, 0.353) | ||
| 7 | chr3q13 | sig chemotaxis | drug resistance | amipathway |
| (0.791, 0.188) | (0.915, 0.094) | and metabolism | (0.759, 0.235) | |
| (0.859, 0.12) | ||||
| 8 | chr12q14 | map00252 alanine and | p53pathway | cskpathway |
| (0.777, 0.344) | aspartate metabolism | (0.857, 0.16) | (0.759, 0.235) | |
| (0.903, 0.281) | ||||
| 9 | chr12p12 | map00910 nitrogen | st fas signaling | map03020 rna poly- |
| (0.770, 0.312) | metabolism(0.903, 0.281) | pathway(0.844, 0.22) | merase(0.749, 0.353) | |
| 10 | chr9q31 | rap down(0.903, 0.281) | ca nf at signalling | map00230 purine |
| (0.763, 0.375) | (0.836, 0.12) | metabolism | ||
| (0.748, 0.324) | ||||
| Rank | Leukemia | Lung Boston | Lung Michigan | |
| 1 | chr14q23 | nfkb reduced | st fas signaling | |
| (1, 0) | (0.79, 0.323) | pathway(0.808, 0.291) | ||
| 2 | chr6q22 | nfkb induced | dna damage signal- | |
| (0.998, 0.083) | (0.77, 0.258) | ling(0.805, 0.279) | ||
| 3 | chr14q11 | map00640 propanoate | crebpathway | |
| (0.998, 0.062) | metabolism(0.762, 0.258) | (0.801, 0.291) | ||
| 4 | chr17q25 | map00340 histidine | il17pathway | |
| (0.998, 0.083) | metabolism(0.745, 0.306) | (0.77, 0.326) | ||
| 5 | chr8p21 | hemo tf list jp | st integrin signaling | |
| (0.996, 0.042) | (0.733, 0.323) | pathway(0.765, 0.326) | ||
| 6 | chr6q21 | map00620 pyruvate | map00240 pyrimidine | |
| (0.996, 0.021) | metabolism | metabolism | ||
| (0.732, 0.371) | (0.76, 0.314) | |||
| 7 | chrXq28 | cr immune function | cr immune function | |
| (0.995, 0.042) | (0.72, 0.355) | (0.751, 0.302) | ||
| 8 | chr3q25 | map00970 aminoacyl trna | st ga12 pathway | |
| (0.993, 0) | biosynthesis(0.71, 0.387) | (0.747, 0.349) | ||
| 9 | chr11q23 | proteasomepathway | tgf beta signaling | |
| (0.992, 0) | (0.707, 0.306) | pathway(0.743, 0.302) | ||
| 10 | chr11 | map00380 tryptophan | raccycdpathway | |
| (0.992, 0) | metabolism(0.705, 0.355) | (0.742, 0.291) |
Table 4: Results of real examples using AUCcv. Top ten gene sets selected by the linear combination coefficients for each data set. AUC and classification error rates via 5-fold cross-validation are shown in parentheses.
| Rank | Gender C1 | Gender C2 | P53 | Diabetes |
| 1 | chrY | testis genes from | badpathway | map00252 alanine |
| (0.031, 0.031) | xhx and netaffx | (0.12, 0.14) | and aspartate meta- | |
| (0.062, 0.062) | bolism(0.235, 0.206) | |||
| 2 | chrYp11 | gnf female genes | g2pathway | mef2dpathway |
| (0.031, 0.031) | (0.125, 0.188) | (0.14, 0.14) | (0.235, 0.235) | |
| 3 | chrYq11 | xinact merged | p53pathway | achpathway |
| (0.031, 0.031) | (0.156, 0.125) | (0.14, 0.18) | (0.265, 0.324) | |
| 4 | chrXp22 | sig regulation of the | drug resistance and | ucalpainpathway |
| (0.156, 0.156) | actin cytoskeleton by | metabolism(0.16, 0.24) | (0.294, 0.324) | |
| rho gtpases(0.281, 0.25) | ||||
| 5 | chr12q15 | st dictyostelium disco- | mitochondriapathway | map03020 rna poly- |
| (0.281, 0.406) | ideum camp chemotaxis | (0.16, 0.14) | merase(0.324, 0.294) | |
| pathway(0.281, 0.219) | ||||
| 6 | chr3q13 | map00051 fructose and | p53 signalling | mprpathway |
| (0.312, 0.281) | mannose metabolism | (0.16, 0.22) | (0.324, 0.353) | |
| (0.312, 0.312) | ||||
| 7 | chrXp11 | map00252 alanine and | p53hypoxiapathway | xinact merged |
| (0.344, 0.281) | aspartate metabolism | (0.16, 0.2) | (0.324, 0.206) | |
| (0.312, 0.344) | ||||
| 8 | chr2q14 | map00910 nitrogen | radiation sensitivity | krebs-tca cycle |
| (0.375, 0.406) | metabolism(0.312, 0.25) | (0.16, 0.18) | (0.353, 0.294) | |
| 9 | chrX | cr dna met and mod | p53 up | map00710 carbon |
| (0.375, 0.312) | (0.344, 0.406) | (0.16, 0.18) | fixation(0.353, 0.324) | |
| 10 | chr5p13 | sig chemotaxis | atmpathway | s1p signaling |
| (0.406, 0.375) | (0.344, 0.219) | (0.18, 0.18) | (0.353, 0.412) | |
| Rank | Leukemia | Lung Boston | Lung Michigan | |
| 1 | chr3q25 | vippathway | cell adhesion molecule | |
| (0, 0) | (0.306, 0.355) | activity(0.244, 0.256) | ||
| 2 | chr8p21 | map00330 arginine | no1pathway | |
| (0, 0) | and proline meta- | (0.244, 0.256) | ||
| bolism(0.339, 0.371) | ||||
| 3 | chr3q21 | proteasome degrad- | nfkb induced | |
| (0, 0.021) | ation(0.339, 0.355) | (0.244, 0.267) | ||
| 4 | chr7p15 | p53 down | hoxa9 down | |
| (0, 0) | (0.339, 0.258) | (0.244, 0.244) | ||
| 5 | chr3 | hox list jp | testis genes from | |
| (0, 0.021) | (0.339, 0.355) | xhx and netaffx | ||
| (0.244, 0.244) | ||||
| 6 | chr11 | downreg by hoxa9 | alkpathway | |
| (0, 0.042) | (0.339, 0.355) | (0.256, 0.267) | ||
| 7 | chrY | atmpathway | at1rpathway | |
| (0, 0) | (0.355, 0.371) | (0.256, 0.267) | ||
| 8 | chr10q24 | calcineurinpathway | map00230 purine | |
| (0.021, 0.021) | (0.355, 0.355) | metabolism(0.256, 0.279) | ||
| 9 | chr8p11 | cell growth and or | map00860 porphyrin and | |
| (0.021, 0.021) | maintenance | chlorophyll metabolism | ||
| (0.355, 0.387) | (0.256, 0.256) | |||
| 10 | chr3p25 | g2pathway | rac1pathway | |
| (0.021, 0) | (0.355, 0.306) | (0.256, 0.267) |
Table 5: Results of real examples using pathwayRF. Top ten gene sets selected by the pathwayRF method for each data set. OOB and 5-fold cross-validation error rates are shown in parentheses.
Figures 5 and 6 describe the within-set total non-zero coefficients of the top five ranking gene sets, where the y-axis labels stand for gene set labels and the x-axis labels consist of the total genes where coefficients are not equal to 0 in these top five gene sets. In these two figures, as before, the up- and down-directions of the short vertical lines represent whether the coefficients are positive or negative, respectively. The lengths of lines stand for the magnitudes of the absolute values of the coefficients. From Tables 4 and 5, we found that there are some gene sets that are simultaneously identified using AUCcv and pathwayRF with OOB, such as Gender (C1), Gender (C2) and P53. However, many gene sets chosen by these two methods are different, such as in the diabetes, and lung cancer data sets from the Boston study and Michigan study. Note that for leukemia data set, there are many gene sets that have excellent discrimination ability; that is, no classification error. In fact, there are many gene sets selected by both of AUCcv and OOB in this case. Table 6 lists the average five-fold cross-validations error rates of the top-ten gene sets identified by AUCcv and pathwayRF, which suggests that the proposed AUCcv is comparable to pathwayRF. The PTIFS method chooses only one gene set in most of the data sets, except the lung cancer data set from the Boston study. The longer the vertical line, the greater the discrimination ability of genes within the gene set. Hence, it is shown in Figures 5 and 6 which genes have the greatest ability to separate the two groups (e.g. male and female, ALL and AML) among those genes under consideration. From a statistical perspective, when the number of variables is much larger than the number of subjects, it is difficult to have a firmly, satisfactory variable selection scheme which is overwhelmingly better than others. Thus, further study on the selected variables is essential. However, in the gene set selection problem discussed here, the selected gene sets have to be re-confirmed through intensive biological laboratory work. Hence, the parsimoniousness of the proposed method is usually preferable in this respect.

Figure 5: Within-set coefficients for real examples. Within-set total non-zero coefficients of the top five gene sets for Gender, P53 and Diabetes data sets.

Figure 6: Within-set coefficients for real examples. Within-set total nonzero coefficients of the top five gene sets for Leukemia, Lung Boston and Lung Michigan data sets.
| Method | Gender C1 | Gender C2 | P53 | Diabetes |
| pathwayRF | 0.231 | 0.237 | 0.180 | 0.297 |
| AUC | 0.193 | 0.156 | 0.138 | 0.317 |
| Method | Leukemia | Lung Boston | Lung Michigan | |
| pathwayRF | 0.012 | 0.346 | 0.260 | |
| AUC | 0.033 | 0.324 | 0.307 |
Table 6: Results of real examples. Average five-fold cross-validation error rates of the top-ten gene sets identified by AUCcv and pathway RF.
We propose a gene set selection method based on the discrimination abilities of gene sets with AUC as the classification performance criterion. This kind of a discrimination-based method is rarely discussed in gene set selection research and is different from methods that are based on clinical outcomes or phenotypes. Our algorithm is founded on the PTIFS algorithm [31], which can select features from high dimensional data sets with small number of subjects. We also introduce two AUC-based statistics to assess the discriminant abilities of gene sets for binary class distinctions. In addition to selecting the gene set, our algorithm can further quantify the impact of individual genes within the gene set when the gene set-based classification of phenotypes is conducted.
From the numerical results of the synthesized data set, we found that the proposed method successfully selects the targeted gene sets. In the numerical results from analyzing real data, the selected gene sets are somewhat different from those selected in other papers that analyse the same data sets based on other association analysis. This difference is primarily due to the fact that the classic gene set-based tests aim to detect significant gene sets in which genes are differentially expressed between phenotypic conditions. However, our method is to identify gene sets with regard to their ability to predict phenotypic conditions. As we know, the amount of enrichment will influence the ability of identifying differential gene sets. The GSEA is conservative, especially when the percentage of differentially expressed genes is relatively small within the gene set. This has been clearly illustrated by applying GSEA to two lung cancer data sets published as supporting information on the GSEA web site, since most gene sets are not statistically significant in terms of p-values. Nonetheless, the simulation studies reveal that our method is able to identify gene sets with small alterations between two phenotypes, even when only 5% of genes in the gene set are differentially expressed. Specifically, we found two nuclear factor-KB (NFKB)-related sets with higher AUC in the Boston Lung cancer data. These gene sets are thought to be major transcription factors regulating many important signaling pathways involved in the tumor promotion. In contrast, there is a large overlap among the significant gene sets between the two methods in the Gender data sets. This result is due to a large proportion of differentially expressed genes within the sets. In summary, our method provides a powerful alternative to gene sets information methods currently available in the literature. The gene sets selected by our approach may reveal distinct prospects of expression profiles, which are useful for biologists when discrimination ability is of concern.
Concerning the framework of multiple-testing issue, the false discovery rate (FDR) approach can be used to adjust for multiple comparisons using p-values derived from two AUC-based statistics. However, the FDR and the estimated q-values depend on the number of gene sets. Since the construction of gene sets is based on the biologically relevant information retrieved from public databases, the number of gene sets may be different across different databases and different gene sets may share common genes. Thus, it is critical to select the thresholds to control for FDR's across different experiments and count for the possible complex correlation structure among p-values. Further work is required in order to estimate error rate and therefore it is not discussed in this article.
There are several possible extensions of the proposed method; for example, we can replace AUC with other performance measures such as partial AUC. In addition, the linear combination within gene sets can be replaced by other methods, even apply a highly nonlinear classification algorithm, if our only concern is to identify gene sets and not the impact of individual genes. In fact, from the prospective of gene sets selection, we do not even require that the classification methods used within gene sets be homogeneous. We can simply allow the classification method used for each gene set to be the best for that particular gene set among a group of classifiers under consideration, and then the rest of the steps can still be easily applied.
This work is partially supported via NSC97-2118-M-001-004-MY2 and NSC98- 2118-M-039-002 funded by the National Science Council, Taipei, Taiwan, ROC, and National Natural Science Foundation of China (Grant No. 11101396).