Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2022)Volume 15, Issue 10
Pathway enrichment analysis assists researchers in gaining mechanical insights into the list of genes generated by genome-scale experiments (omics). In this work, we used breast cancer gene expression data to recognize genetic pathways. The pathways were conducted based on KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO-ALL (Gene Ontology) using the PathfindR tool. The gene expression data were downloaded from the Pan-Cancer-Atlas using the R studio program. Preprocessing steps were performed on the downloaded gene expression data. These steps are as follows: First, the outlier samples were removed second; a normalization process was applied to the data. Third, a filtering process is applied to the data. DESeq2 package is used to find Differentially Expressed Genes (DEGs).Thereafter, we used the pathfindR software to conduct the enrichment analysis .also, and we construct a Protein-Protein Interaction network in order to detect active sub networks. The results indicated that there are 73, 63 top pathways that are associated with our Differentially Expressed Genes on GO and KEGG pathways respectively. Moreover, the top genes related to our BRCA include NUP214 NUP62, NUP93, SUMO3, EIF2B1, EIF4A3, RNPS1, and SRRM1.
RNA-Seq; Gene expression; Active subnetwork; PathfindR; Go-All; KEGG pathways
Gene expression profiles from tissue samples can now be generated at a reasonable cost because of recent improvements in DNA gene expression technologies. Gene expression analysis assists scientists and medics in better understanding disease causes and developing platforms that aid in the diagnosis, prognosis, and treatment [1]. Breast cancer is the most common cancer in women around the world. There are several significant risk factors for breast cancer, including older age, null parity, obesity, smoking, and estrogen exposure. In almost 10% of cases of breast cancer, the patients’ have a strong genetic predisposition [2]. Based on the stage of cancer and the presence of lymph nodes, the initial treatment of breast cancer usually involves surgical resection of the tumor [3]. Early detection with appropriate treatment could reduce death rates significantly in the long term, according to previous studies [4]. In addition to mammography and magnetic resonance imaging, ultrasounds, positron emission tomography, and biopsy, researchers have examined several other breast diagnostic methods [5]. The traditional methods have some limitations; these limitations are, less effective for subjects under 40 years of age and with dense breasts, less sensitive to small tumors, not providing an indication of the potential outcome of disease [6,7]. And expensive and involving high levels of radiation [8].
Some researchers they used pathway enrichment analysis approaches include the work of Reimand et al. that conducted analysis of raw RNA-seq data from ovarian cancer samples to define a ranked gene list [9]. moreover the work of Yang et al. analysis using GO and the KEGG pathways enrichment using the Database for Annotation, Visualization and Integrated Discovery(DAVID) software on the gene expression profile dataset GSE26440, In addition, they used the Gene Set Enrichment Analysis approach for enrichment analysis of the DEGs. Their results indicated, the top 10 hub genes, which are all up regulated in septic shock children [10]. Li et al. conducted GO and KEGG enrichment analyses by DAVID and KOBAS database. Their results identified, a total of 134 DEGs by differential expression analysis, consisting of 88 up- and 46 down-regulated genes. GO and KEGG analyses showed enriched terms and pathways related to cell adhesion, tumorigenesis, and cellular immunity. The PPI network identified six hub genes containing CD3D, CD3E, CD3G, FYN, GRAP2, and ITK [11].
Wei et al. performed GO and KEGG enrichment analysis on GSE86374 micro-expression matrix chip data that consist of 159 samples (124 normal samples and 35 breast cancer samples), they obtained 173 up-regulated genes and 143 down-regulated genes for GO and KEGG enrichment analysis. They noted these genes are also significantly enriched in the KEGG pathway, including phenylalanine metabolism, staphylococcus aureus infection, and the PPAR signaling pathway [12]. Chen et al. were performed GO analysis and KEGG pathway enrichment analyses of DEGs by the DAVID on The gene expression profiling (GSE86945, GSE86946 and GSE102088) data, their result identified a total of 2998 DEGs between TNBC and health breast tissue, including 411 upregulated DEGs and 2587 down-regulated DEGs , GO analysis results showed that down-regulated DEGs were enriched in gene expression (BP), extracellular exosome (CC), and nucleic acid binding, and up-regulated were enriched in chromatin assembly (BP), nucleosome (CC), and DNA binding (MF), KEGG pathway results showed that DEGs were mainly enriched in Pathways in cancer and Systemic lupus erythematosus and so on [13]. Deng et al. were conducted the GO and KEGG analysis on four datasets (GSE21422, GSE29431, GSE42568, and GSE61304) from Gene Expression Omnibus (GEO) through FunRich, and their results recognized, 203 up-regulated and 118 down-regulated DEGs and remain Mitotic cell cycle and epithelial-to-mesenchymal transition pathway as major enriched pathways for the up-regulated and down-regulated genes, respectively [14]. Lv et al. were conducted GO and KEGG enrichment analyses on four datasets GSE5847, GSE22597, GSE23720, and GSE45581 downloaded from the Gene Expression Omnibus (GEO) to understand the potential bio-functions of the DEGs. Their results indicated a total of 114 DEGs were identified from the GEO datasets GO and KEGG analyses showed that the DEGs were mainly enriched in oncogenesis and cell adhesion [15]. Hu et al. performed an extreme case-control study including 208 breast cancer patients with poor invasive disease-free survival (iDFS) and 208 patients with favorable iDFS were individually matched on molecular subtype from the Breast Cancer Cohort at West China Hospital (WCH; N=192) and The Cancer Genome Atlas, their result revealed differential expression of genes in the glucocorticoid pathway in tumor tissue (P=0.028), but not normal tissue (P=0.701), was associated with poor iDFS, Somatic mutation of the adrenergic and cholinergic pathways was significantly associated with iDFS in WCH, but not in TCGA. And also were discovered the glucocorticoid pathway may play a role in breast cancer prognosis through differential mutations and expression. [16].
From the previous study, however, how these pathways interact between genes remains unclear. In the present study, the gene expression data were downloaded from the Pan-Cancer-Atlas using “Illumina HiSeq” platform on R software. Preprocessing steps were performed on the downloaded data. These steps are as follows: First, the outlier samples were removed second, a normalization process was applied to the data, and third a filtering process is applied to the data. We conducted a differential expression analysis using R DESeq2 program to create a list of efficient genes prioritized by DEGs scores, also we used the pathfindR tool, to construct Protein-Protein Interaction that contains interaction information between the genes and finds active subnetworks to complete the enrichment analysis with GO and KEGG Pathways Beyond differential expression analysis of breast cancer gene expression data for discover top pathways and genes associated with DEGs that affected Breast Cancer (BRCA). Our result identify 73and 63 as top pathways and sub sequentially the top genes in each on GO and KEEG pathways enrichment analysis respectively.
Dataset
Gene expression data sets of breast-cancer (BRCA) were downloaded from Pan-Cancer Atlas [17] using the R studio program. The Genomic Data Commons (GDC) is an acronym for the National Cancer Institute’s Genomic Data Commons, which offers the research group with an integrated data store that allows data sharing through cancer genomic investigations. The GDCquery function requires input of several parameters. The project case denotes that the project has to be selected from the Pan-Cancer Atlas’s list of legal projects. The value of this case in terms of breast cancer will be “TCGA-BRCA [18]. The legacy option can be set to true or false; we chose true, indicating that the query should be included in the inheritance database when we appeal the data. The inheritance repository shall create an unchanged copy of data that was previously available for download on the TCGA Data Portal. Every project has its own data category; therefore data.category refers to the type of data in the project. We need to change the data category to “Gene expression” in our case. The data Type option defines the type of data that will be used to filter the files that will be downloaded. We set the data type to “Gene expression quantification” in our example. Genes can be calculated using RNA-Seq to determine the number of reads that map to each gene [19,20]. In terms of the plan, we have options of selecting one of several platforms, so we have chosen “Illumina HiSeq” platform. The “file.ty pe” argument specifies the type of file that can be utilized in a legacy database. There are a variety of experimental methodologies to choose from, such as RNA-Seq, miRNA-Seq, and Genotyping Array. In our case, RNA-Seq was used to build our expression profile [21,22], for example. Finally, the sample type specifies the kind of sample that can be used to filter the data that will be downloaded. In our situation, the sample type can be set to “c (“Primary solid Tumor’ ‘Solid Tissue Normal”),” which means that the gene expression of ordinary cases and cases with a tumor compared .The downloaded BRCA data is transformed into a matrix formula. The columns in this matrix indicate the samples or instances, whereas the rows reflect the genetic ranges of interest [23,24]. There are 1208 clinical samples in the BRCA database, with a total of 19948 genes. Because of the enormous number of genes, the data will be sensitive to noise as a consequence of the big number of genes, and the analysis’ performance may be affected. As a result, preprocessing procedures are used to the BRCA data in order to reduce the number of genes and then choose those that contribute significantly in discovering breast cancer genes.
Data preprocessing
To detect problematic arrays, the preprocessing stage was created in this research by building a symmetric square matrix of Spearman correlation across data. Samples that are regarded as outliers are deleted based on this symmetric matrix. Outliers are defined using a correlation threshold of 0.6, and then the BRCA gene expression data is normalized to appropriately estimate expression levels from the BRCA gene expression data, confirming that expression measurement bias can be avoided [25- 28]. The normalizing step is carried out using the TCG Analysis Normalization function from the TCGA biolinks package. GC content is the mechanism used in this role; this is the number of nucleotides in a nucleic acid chain that inhibits either guanine (G) or cytosine (C) in the nucleic acid chain (C). Consequently, the normalization procedure eliminates the reliance on the GCricher gene being accurately DE, as well as the substantial C Content bias [25]. We have completed preprocessing of the data by filtering the gene expression dataset with a threshold of 0.25. H. To select the average value for all samples greater than 0.25.
Differentially expressed genes (Degs)
The R DESeq2 package was used to do a differential expression analysis. Using a significance level of 0.01 in this phase. A generalized linear model (GLM) is used by the DESeq2 differential expression analysis program. The form generalized linear model (GLM) is:
gij~ NB (μij,αi)
μij = sjqij
log2 (qij) = xj.βi
Here gij denotes the amount of Gene i in sample j. These numbers are computed using a Negative Binomial Distribution with an adaptive mean μij and a gene-specific variance factor αi. The adjusted average is translated to a factor qij, which is proportional to the sample-specific size factor c and the estimated true attention of sample j fragment. For each column of the model or design matrix X, the factor βi represents a log2 fold change in Gene i.
The sample and gene-dependent normalization factor sij can be used to generalize the model. The diffusion parameter αi defines the connection between the variance of the observed counts and their mean. In other words, the size factor sj and the covariate dependent component qij, both stated above, decide how far you may extrapolate the observed number from the average value. As a result, the variance function is as follows:
var (gij) = [ gij – μij ] = μij+ αiμij ²
The steps achieved by the DESeq function in the DESeq2 package are the approximation of sj and αi, and the adaptation of the negative binomial GLM to βi and Wald statistics using nbinomWaldTest. Since then, we have calculated the Count Per Million as
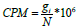
gi Denotes the count observed from the gene i of interest and N is the number of sequenced fragments [29].
Enrichment analysis
We used PathfindR tool to conduct an enrichment analysis with KEGG and GO-ALL Pathways (the representation of the analysis is shown in Figure 1. PathfindR( is a biological grouping function that utilizes a list of genes and related p-values as input values to discover active Subnetworks within a Protein-Protein Interaction Network ), PathfindR also has the ability to group the enriched terms into clusters and select illustrative terms within each cluster [30].In our work we used the gene name and p_value that resulted from differential expression analysis on gene expression data as input to PathfindR. In a Protein-Protein Interaction Network (PPIN), active subnetworks are detected using a list of genes and their associated p values. After that, we performed enrichment analysis on the identified active-subnetworks in order to find enriched terms which might explain the attention phenomenon. The active subnetworks are those within PPIN (by default, BioGRID) that have the highest score (based on the significant difference value specified). These subnetworks are distinct sets of interacting genes that are related to a certain disease and were discovered either during the investigation or as a result of strong gene interaction. PathfindR maps each input gene against a PIN after passing out input (a p-value threshold of 0.01 was used to exclude differential expression data) (Figure 2) [31]. The mapped genes are used to conduct an active subnetwork search (by default, using the greedy method). The active subnetworks that arise are filtered according to the marks, and as a result, the set of relevant genes they cover. The enrichment analysis (hypergeometric distribution test overrepresentation analysis) is then performed on the filtered list of active subnetworks. That is, each gene in the active subnetworks is used to identify highly enriched pathways.

Figure 1: Flow-chart of enrichment analysis with KEGG and GO-ALL Pathways.

Figure 2: It is showing flow-chart of the path find R method steps in detailed manner.
Identification of differentially expressed genes
We used RNA-Seq gene expression profile to find the top pathways and genes of breast cancer. In our study, first; we downloaded RNA-Seq gene expression profiles of breast-cancer using The Cancer Genome Atlas (TCGA) database, which contains 1208 clinical samples with 19948 genes. And preprocessed it, after preprocessing, we got 1208 clinical samples with 14477 genes, including 113 negative samples and 1095 positive samples. Second, we used R software to do a differential expression analysis on these datasets, and we found 3,676 DEGs in the gene expression data using the 0.01 significant threshold levels, as shown in Table 1 as sample from a differentially expressed genes results, the complete results is in APPNDEX1.
| Gene name | logFC | logCPM | PValue | FDR |
|---|---|---|---|---|
| HAS1 | -2.782362 | 0.89763 | 5.29E-218 | 2.50E-216 |
| NMU | 5.790262 | 0.979308 | 1.53E-217 | 7.20E-216 |
| GATA4 | 5.897582 | 0.616554 | 7.91E-217 | 3.72E-215 |
| HLF | -2.761301 | 3.774738 | 8.34E-217 | 3.91E-215 |
| CCDC3 | -2.758493 | 4.817181 | 1.69E-216 | 7.88E-215 |
| KLHL29 | -2.760565 | 2.68592 | 3.45E-216 | 1.61E-214 |
| C7orf58 | -2.752138 | 4.019433 | 3.66E-215 | 1.70E-213 |
| SLC22A3 | -2.755464 | 1.81441 | 1.73E-214 | 7.98E-213 |
Table 1: Sample from Results of DEGs.
KEGG and GO pathway enrichment analysis
We used PathfindR program to complete Enrichment analysis with KEGG and GO-ALL Pathways. a list of genes and related p-values that outperformed from differential expression analysis on gene expression data of breast cancer was utilized as input value to PathfindR to discover active Subnetworks within a Protein-Protein Interaction Network and then KEGG and GOALL pathway enrichment analyses were performed on these active Subnetworks. Enrichment analysis is performed using the genes in each of the active subnetworks, the lowest adjusted p-value for each term (entire active subnetworks) is reserved, and enriched pathways with adjusted p-values more than the certain threshold (0.02) are excluded, this process is known as “Active Subnetwork Search Enrichment Analysis” and it is frequently carried out in parallel for a set number of repetitions (default is 10). For each considerably enriched pathway, the total number of events is also given, with a p-value that is always low and therefore optimally tuned across all iterations. We obtained 63 and 73 as top pathways and subsequently the top genes in each on KEEG and GO pathways enrichment analysis respectively. Samples of the results are displayed in Table 2 and Table 3 for KEEG and GO respectively because the results were featured in form of HTML files and too many to be shown the full results of the pathways are presented in APPNDEX2 for KEEG and APPNDEX3 for GO. Each pathway contains “Term_Description” (represent enriched term explanation), “Fold_Enrichment” (represent Enriched term value), “lowest p-value” (adjusted minimum p-value for the term specified with all rounds), the “highest p-value “for all iterations (adjusted maximum p-value for the term specified with all rounds), “Up-regulated” (upregulated genes for inputs separated by commas in the gene set for the specified term) and “Downregulated” (downregulated genes for inputs separated by commas in the gene set for the specified term). The first KEGG pathway from the top ten pathways includes (NUP214, NUP62, NUP93, SUMO3, EIF2B1, EIF4A3, RNPS1, and SRRM1) and the first GO from the top ten pathways includes (BUD31, LSM3, SF3B6 HNRNPD, HNRNPH1, PCBP1, PTBP1, SRSF5, EIF4A3, HNRNPR, SRRM1, RNPS1, SF3B2, U2AF2, PUF60). The above-mentioned genes and the remains genes of pathways have a significant effect on breast cancer. Table 4 contains converted gene symbol (the gene symbols that are found in the input but not found in the PIN is converted to an alias symbol found in the PIN) and Table 5 contains genes without Interactions (genes that are not found in the PIN) APPNDEX6 contains the full results of genes without Interactions. Sample of visualization of resulted pathways presented in Figure 3 for KEEG pathways, the overall graphs that explain the pathways at 10 iterations are presented in APPENDEX4 and Figure 4 display sample of the protein-protein interaction on GO-ALL pathways, while APPNDEX5 contains all protein-protein interaction of the network.
| ID | Top-Pathway | Fold-Enrichment | Lowest-p | Highest-p | Up-regulated | Down-regulated |
|---|---|---|---|---|---|---|
| hsa03013 | RNA transport | 3.182691 | 1.90E-05 | 9.40E-04 | NUP214 | NUP62, NUP93, SUMO3, EIF2B1, EIF4A3, RNPS1, SRRM1 |
| hsa04966 | Collecting duct acid secretion | 5.355489 | 2.70E-05 | 2.70E-05 | ATP6V1D, ATP6V0E1 | |
| hsa05166 | Human T-cell leukemia virus 1 infection | 3.179058 | 3.80E-05 | 3.80E-05 | TRRAP, NFATC3, IL2RB, JAK1, VDAC1, SLC25A5, ZFP36, SRF, ETS1, CREB1 | |
| hsa03040 | Spliceosome | 4.321326 | 4.20E-05 | 4.20E-05 | SF3B6, LSM3, BUD31 | SF3B2, U2AF2, PUF60, EIF4A3, PCBP1, SRSF5 |
| hsa00410 | beta-Alanine metabolism | 4.641424 | 4.20E-05 | 4.20E-05 | CNDP2, ALDH9A1 | |
| hsa00020 | Citrate cycle (TCA cycle) | 6.962136 | 4.20E-05 | 1.20E-04 | MDH2, PDHA1, PDHB | |
| hsa04921 | Oxytocin signaling pathway | 3.248997 | 4.80E-05 | 4.80E-05 | MYL6B, MYL6 | EEF2K, EEF2, CALM3, NFATC3, ACTG1 |
| hsa05203 | Vira carcinogenesis | 2.953633 | 7.00E-05 | 7.00E-05 | GTF2B | CREB1, JAK1, RBL2, HDAC1, DNAJA3, SRF |
| hsa04210 | Apoptosis | 1.547141 | 9.30E-05 | 9.30E-05 | DDIT3 | ACTG1, PARP1 |
| hsa00620 | Pyruvate metabolism | 7.140652 | 9.40E-05 | 9.40E-05 | PDHA1, PDHB, ALDH9A1, MDH2 |
Table 2: Top ten KEGG pathway based on the p-value.
| ID | Top-Pathway | Fold-Enrichment | Lowest-p | Highest-p | Up-regulated | Down-regulated |
|---|---|---|---|---|---|---|
| GO:0000398 | mRNA splicing, via spliceosome | 5.069516 | 2.60E-09 | 5.80E-09 | BUD31, LSM3, SF3B6 | HNRNPD, HNRNPH1, PCBP1, PTBP1, SRSF5, EIF4A3, HNRNPR, SRRM1, RNPS1, SF3B2, U2AF2, PUF60 |
| GO:0016070 | RNA metabolic process | 9.945908 | 4.90E-07 | 1.60E-04 | HNRNPD, HNRNPH1, PCBP1, PTBP1, HNRNPR, DDX54 | |
| GO:0006406 | mRNA export from nucleus | 6.459714 | 3.10E-06 | 8.10E-05 | NUP214 | SRSF5, SLBP, NUP93, EIF4A3, SRRM1, RNPS1, U2AF2, NUP62 |
| GO:0008134 | transcription factor binding | 3.238203 | 3.30E-06 | 3.30E-06 | DDIT3, GTF2B, HMGB2 | PARP1, GATA2, HDAC1, SMARCA4, SRF, BRD7, MDFIC |
| GO:0043968 | histone H2A acetylation | 10.710978 | 2.50E-05 | 2.50E-05 | TRRAP, EP400 | |
| GO:0006974 | cellular response to DNA damage stimulus | 3.010653 | 2.80E-05 | 2.80E-05 | DDIT3, TRIP12, CBX3 | ABL1, PARP1, RAD17, RPA1, HELB |
Table 3: Top six GO pathways based on the p-value.
| Old Symbol | Converted Symbol | Change | p-value | |
|---|---|---|---|---|
| d131 | BICDL1 | CCDC64 | -0.65054 | 0.0058223 |
| 206 | NRDC | NRD1 | 0.4313611 | 0.0094019 |
| 306 | ADGRG5 | GPR114 | -0.487876 | 0.0183865 |
Table 4: Table of converted gene symbols.
| Gene No. | GeneSymbol | Logfc | adj.p-val |
|---|---|---|---|
| 29 | MFNG | -0.3848755 | 0.0009377 |
| 95 | LOC101929243 | 0.3482631 | 0.0039567 |
| 265 | S100A12 | 0.8084613 | 0.014864 |
| 273 | SLC11A1 | 0.5612296 | 0.0154375 |
| 314 | USP11 | -0.2560524 | 0.0202169 |
| 315 | MTMR12 | -0.269036 | 0.0202169 |
| 316 | CRLF3 | -0.3244654 | 0.0202744 |
| 317 | SYPL1 | -0.2851805 | 0.0210506 |
| 318 | JMJD8 | -0.3762269 | 0.0212812 |
| 319 | CRELD2 | -0.3054072 | 0.0212812 |
| 320 | DDOST | -0.3388948 | 0.0213069 |
| 321 | UBE2G1 | -0.3992315 | 0.0213069 |
| 322 | XYLT2 | -0.3123587 | 0.0218339 |
| 323 | WDR33 | -0.3197996 | 0.0218339 |
Table 5: Table of Genes without Interactions (not found in the PIN).

Figure 3: Explains the visualization of enriched terms of KEEG pathway enrichment analysis.

Figure 4: It shows the protein _protein interaction network, the green nodes indicate the up regulated genes, while the red nodes indicate
the down regulated genes.
PathfindeR was used to completing the enrichment analysis by using gene names and their p-values(which outperformed from a differential expression analysis on gene expression data of 1208 clinical samples of 19948 genes of breast cancer (BRCA) downloaded from Pan-Cancer Atlas by R studio software, we achieved 3,676 DEGs) as input value to identify active Subnetwork in Protein-Protein Interaction Networks to perform the enrichment analysis for these active networks (which contains a list of important genes) with the GO-ALL and KEGG pathways. From the results of the analysis, we achieved the top pathways and consequently top genes that affect breast cancer disease. In a comparison of the results of our study with others for example the study of Li et al. performed GO and KEGG enrichment analysis to discover Hub Genes and Pathways associated with Triple Negative Breast Cancer (TNBC) Based on Expression Profiles Analysis using DAVID and KOBAS. Built on differential expression analysis of four TNBC datasets, a total of 134 DEGs were achieved. The enriched terms and pathways were related primarily to cell adhesion, cancer, and cellular immunity, according to GO and KEGG analysis. Then they build a PPI network, the PPI network discovered six hub genes, counting CD3D, CD3E, CD3G, FYN, GRAP2, and ITK [12]. But, in order to identify enriched terms, our study first detects active sub networks in a protein-protein interaction network and then performs enrichment analysis on the recognized sub-networks. Our result of analysis shows (63 and 73 as top pathways and sub sequentially the top genes found in each pathway on KEEG and GO-ALL enrichment analysis respectively), compared to their study we found the results of our analysis show more information about the interaction between the genes mentioned in Figure 4.
We identify the top pathways and genes that affect breast cancer using RNA-Seq gene expression profiles. Using R studio, we downloaded gene expression profiles from Pan Cancer Dataset. The data is downloaded using “Illumina HiSeq”. Outlier samples are removed by preprocessing the downloaded data, as defined by AAIC, which is the matrix of Spearman coefficients between samples. We appropriately calculated the expression level and avoided biases in the expression measurements by adding a normalization method to the data. The data is then filtered. DEGs were identified in breast cancer Gene Expression Data; our findings indicate that DEGs can be used to identify the top pathways of breast cancer. Using the differential expression analysis result (gene names and p-values), we used PathfindeR for identifying active Sub network in protein-protein interaction networks and complete enrichment analysis for these active networks with the GO-ALL and KEGG pathways. Several useful top pathways and top genes that are effective in breast cancer disease have been identified. We recommend in the future; further experiments will need to be done to determine if DEGs affect one or more DEG signaling pathways depending on their expression level, function, and effect.
This study did not receive any specific funding from any public sector or nonprofit funding agency.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Cross ref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Abdeljalil EM, Elbashir MK, Akode AO (2022) Identification of Breast Cancer Pathways Based on Gene Expression Data. J Proteomics Bioinform. 15:612
Received: 26-Oct-2022, Manuscript No. JPB-22-19851; Editor assigned: 31-Oct-2022, Pre QC No. JPB-22-19851 (PQ); Reviewed: 14-Nov-2022, QC No. JPB-22-19851; Revised: 21-Nov-2022, Manuscript No. JPB-22-19851 (R); Published: 28-Nov-2022 , DOI: 10.35248/0974-276X.22.15.612
Copyright: © 2022 Abdeljalil EM, et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.