Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2009) Volume 2, Issue 11
Recent studies have shown that monoterpenes exhibit antitumor activities and suggest that these compounds are a new class of cancer chemo-preventive agents. Limonene, a main constituent of orange and citrus peel oils has been reported to exert antitumor activity against mammary gland, lung, liver, stomach and skin cancers in rodents whereas, geraniol, a principal constituent of Geranium and Ocimum inhibits the growth of human colon cancer cells. Prenylation of proteins is essential for progression of cells into the S phase and involves post-translational covalent attachment of a lipophilic farnesyl or geranylgeranyl isoprenoid group to numerous proteins. Suppression of prenylation of proteins leads to inhibition of DNA synthesis. Further, epidemiologic evidences suggest that suppression of hydrophilic 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, a key enzyme of mevalonate biosynthesis, leads to reduction of the mevalonate pool and thus limits protein isoprenylation. Geraniol and limonene inhibit the activity of HMG-CoA reductase subsequently reducing the possibility of cancer growth. In the present work, we analyzed binding affinity of limonene and geraniol with HMG-CoA and explored mechanism of interaction using in silico approaches. The binding positions were verified according to their energy, PMF (Potential of Mean Force) value, PLP (Piecewise Linear Potential) value and Ligand Internal energy. It was found that limonene had greater binding affinity with the receptor suggesting better antitumor agent in comparison to geraniol.
Keywords: Limonene, Geraniol, HMG CoA reductase, Cancer chemo-preventive agents, docking.
Essential oils are highly concentrated volatile aromatic essences of plants. They are mainstay of aromatherapy but are also used in flavoring, perfumes and even as solvents. Terpenes, aldehydes, esters, ketones, alcohol, phenol and oxides are major components of essential oils. Monoterpenes function physiologically as chemo-attractants or chemo-repellents, and they are largely responsible for the distinctive fragrance of many plants (McGarvey et al., 1995). Significant scientific evidences are there to suggest that nutritive and non-nutritive plant-based dietary factors can inhibit the process of carcinogenesis effectively (Singletery, 2000). Monoterpenes are non nutritive dietary components found in the essential oils of plants having antitumor activity, exhibiting not only the ability to prevent the formation or progression of cancer, but also regress existing malignant tumors (Crowell, 1999). The human exposure to monoterpenes through the diet or environment is widespread.
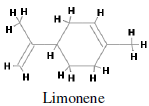
Major monoterpenes includes limonene, pinenene, menthol, geraniol, camphene, sabinene, cadinine. Monoterpenes consist of two isoprene units with the molecular formula C10H16. Monoterpenes may be linear (acyclic) or contain rings. These 10 carbon isoprenoids are derived from the mevalonate pathway in plants but are not produced by mammals, fungi or other species (Loza-Tavera, 1999). Citrus fruit, orange and peppermint are the main sources of d-limonene i.e. p-mentha-1,8-diene (Kodama et al., 1977). d-limonene (Figure 1) is a prevalent flavoring agent and because of its pleasant citrus fragrance, it is commonly added to cosmetics, soa psand other cleaning products. It is a cyclic monoterpene and formed by the cyclization of geranylpyrophosphate in a reaction catalyzed by limonene synthase (Alonso et al., 1992; Kjonaas et al., 1983). Limonene has well-established chemo-preventive activity against many cancer types. Limonene has been shown to inhibit the development of spontaneous neoplasms in mice at the dose of 1200 mg/kg orally (National Toxicology Program, 1990). Dietary limonene also reduces the incidence of spontaneous lymphomas in p53-/- mice (Salim et al., 2003). When administered either in pure form or as orange peel oil (95% d-limonene), limonene inhibits the development of chemically induced rodent mammary (Asamoto et al., 2002), skin (Elegbede et al., 1986), liver (Lu et al., 2004), lung and stomach (Raphael and Kuttan, 2003) cancers. In rat mammary carcinogenesis models, the chemo-preventive effects of limonene are evident during the initiation phase of 7-12- dimethylbenz[a]anthracene (DMBA)-induced cancer (Elson et al., 1988) and during the promotion phase of both DMBA- and nitrosomethylurea (NMU)-induced cancers (Chander et al., 1994). Dietary limonene also inhibits the development of ras oncogene–induced mammary carcinomas in rats (Gould et al., 1994). Development of azoxymethane-induced aberrantcrypt f oci in the colon of rats was significantly reduced when they were given 0.5% limonene in the drinking water (Kawamori et al., 1996).

Figure 1: Chemical structure of d-limonene (p-mentha-1,8-di-ene ).
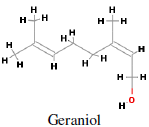
Main sources of geraniol i.e. trans-3,7-Dimethyl-2,6-octadien- 1-ol (Figure 2) are bergamot, carrot, coriander, lavender, lemon, lime, nutmeg, orange, rose, blueberry, basil and blackberry. It is mainly used in perfumery and flavouring industries. Geraniol synthase is involved in the terpene biosynthetic pathway converting geranyl diphosphate to geraniol (Iijima et al., 2004). Geraniol, an acyclic monoterpene, has antitumor activity against mu rine leukemia, hepatoma and melanoma cells in vivo when administered before and after tumor cell transplantation. It has antiproliferative effects on hepatoma and melanoma cell growth (Polo and de Bravo, 2006). Geraniol (400 µM) caused a 70% inhibition of cell growth in human colon cancer cell lines. Geraniol has shown anti-tumoral efficacy on TC-118 human tumors transplanted in Swiss nu/nu mice. Geraniol (150 µM) has been identified to reduce thymidylate synthase and thymidine kinase expression in cancer cells. In nude mice, the combined administration of 5-fluorouracil (20 mg/kg) and geraniol (150 mg/kg) caused a 53% reduction of the tumor volume, whereas a 26% reduction was obtained with geraniol alone (Carnesecchi et al., 2004).

Figure 2: Chemical structure of geraniol (trans-3,7-Dimethyl-2,6-octadien-1-ol).
HMG-CoA reductase (Figure 3) is a polytopic, transmembrane protein that catalyzes a key step in the mevalonate pathway (conversion of HMG-CoA to mevalonate). Mevalonate is necessary for cell growth (Swanson and Hohl, 2006) and is involved in the synthesis of sterols, isoprenoids and other lipids. HMG-CoA reductase is the rate-limiting step in cholesterol synthesis and represents the sole major drug target for contemporary cholesterol- lowering drugs (Genser et al., 2008). HMG-CoA reductase is also an important developmental enzyme. Limonene and geraniol suppress HMG-CoA reductase synthesis in mammalian cells by decreasing the translational efficiency of HMG-CoA reductase transcripts (Peffley and Gayen, 2003) and thus reduce mevalonate production. Terpenoids reduce cancer formation by the simple reduction of synthesis of chlolesterol and ubiquinone and other cholesterol derivatives that are necessary for the cell proliferation. It is speculated that mevalonate is probably involved in the post-translational modification of proteins involved in cell turnover. The reduction of the mevalonate pool limits protein isoprenylation, which involves the post-translational covalent attachment of a lipophilic farnesyl or geranylgeranyl isoprenoid group to numerous proteins (Clarke, 1992).

Figure 3: Rotatable bonds shown in green circle (A) Geraniol: with 5 rotatable bonds (B) Limonene: with 1 rotatable bond.
Preparation of the Receptor and the Ligands
The structure file of HMG-CoA reductase complexed with atorvastatin (an inhibitor) was downloaded from Protein Data Bank (PDB id: 1HWK). Structure was resolved using x-ray crystallography experiment at 2.22 Å resolution with R-value 0.212 from Homo sapiens (Istvan and Deisenhofer, 2001). To study the interaction of HMG-CoA reductase with geraniol and limonene, water molecules and non-protein residues were deleted from the complex. CHARMm forcefield was applied to study the molecular dynamics. CHARMm uses a flexible and comprehensive empirical energy function that is a summation of many individual energy terms. The energy function is based on separable internal coordinate terms and pairwise nonbond interaction terms (Brooks et al., 1983). The total energy is expressed by equation 1.
E = Eb + Eθ + Eφ + Eω + EvdW + Eel + Ehb + ECr + ECφ (1)
where, E is the total energy; Eb(bond potential), Eθ(bond angle potential), Eφ(Dihedral angle potential), Eω(improper torsions) are internal energy terms, EvdW(Van der Waals interactions), Eel(Electrostatic potential), Ehb(hydrogen bond energy) are nonbonded internal/external interactions energy terms, ECr(constraints) and ECφ(user defined energy function) are special energy terms.
Identification of Binding Cavity on Receptor Surface
After energy minimization, the binding pockets of the receptor were determined by using “eraser” algorithm using Accelrys Discovery Studio. This algorithm is first used to remove all grid points outside the receptor. The boundary between the “inside” and “outside” region is determined by the “site opening” parameter. For the remaining grid points (i.e., those “inside” the site), a flood-filling algorithm is employed to find contiguous regions consisting of unoccupied, connected grid points. Each such region is identified as a possible site. A user-specified size cutoff used to remove sites smaller than the specified volume for further consideration (Venkatachalam et al., 2003).
Interaction Protocol and Scoring Functions for Docking
The interaction of the receptor and the ligand was performed using “LigandFit” protocol on Accelrys Discovery Studio. In the first phase of LigandFit docking procedure, binding sites were indentified on the receptor surface. Site partitioning approach was followed to sample different parts of the larger binding site for docking. In the second phase, docking between receptor and ligand was performed in the specified site.
Docking ligands to the specified sites has different approaches like conformational search to generate candidate ligand conformations for docking, ligand/site shape matching to select ligand conformations that are similar to the shape of site or site partitions. Candidate ligand poses in the binding site are evaluated and prioritized according to the DockScore function on the basis of forcefield approximation (equation 2), Piecewise Linear Potential function (PLP) (equation 3), LigScore1, LigScore2, Potential of Mean Force (PMF) and Jain scores.
DockScore(forcefield) = - (ligand/receptor interaction energy + ligand internal energy) (2)
DockScore(PLP) = - (PLP potential) (3)
As shown in Eq. 2, this version of DockScore contain two energy terms, these are internal energy of the ligand and the interaction energy of the ligand with the receptor. The interaction energy is taken as the sum of the van der Waals energy and electrostatic energy. To reduce the time needed for the computation of the interaction energy, a grid-based estimation of the ligand/receptor interaction energy is employed. PLP is a fast, simple, docking function that has been shown to correlate well with protein-ligand binding affinities. PLP scores are measured in arbitrary units, with negative PLP scores reported in order to make them suitable for subsequent use in consensus score calculations. Higher PLP scores indicate stronger receptor-ligand binding (larger pKi values). LigScore1 is a scoring function for predicting receptor-ligand binding affinities. vdW, C+pol and TotPol^2 descriptors are used to calculate LigScore1 (equation 4, 5), which is computed in units of pKi (-log Ki). When scoring ligands, the individual contributions of these descriptors may also be provided along with the overall LigScore1 value. Two slightly different equations are used in the calculation of LigScore1 depending on the forcefield (Dreiding or CFF) employed for the calculation of the vdW descriptor and the corresponding charge model (Gasteiger or CFF) used to assign atoms as polar or nonpolar.
LigScore2 is another fast and simple scoring function for predicting receptor-ligand binding affinities. vdW, C+pol, and BuryPol^2 descriptors are used to calculate LigScore2 (equation 6,7), which is computed in units of pKi (-log Ki). When scoring ligands, the individual contributions of these descriptors may also be provided along with the overall LigScore2 value. Two slightly different equations are used in the calculation of LigScore2 depending on the forcefield (Dreiding or CFF) employed for the calculation of the vdW descriptor.
LigScore1_CFF = 0.4896 - 0.04551*vdW + 0.1439*C+pol - 0.001010*TotPol^2 (4)
LigScore1_Dreiding = -0.3498 - 0.04673*vdW + 0.1653*C+pol -0.001132*TotPol^2 (5)
LigScore2_CFF = 1.900 - 0.0730*vdW + 0.06246*C+pol - 0.00007324*BuryPol^2 (6)
LigScore2_Dreiding = 1.539 - 0.07622*vdW + 0.6501*C+pol - 0.00007821*BuryPol^2 (7)
where the coefficients were obtained through regression analysis of the binding affinities of a series of protein-ligand complexes (Krammer et al., 2005).
The PMF scoring function (Muegge et al., 2005) is based on statistical analysis of the 3D structures of protein-ligand complexes. They were found to correlate well with protein-ligand binding free energies while being fast and simple to calculate. The scores are calculated by summing pairwise interaction terms over all interatomic pairs of the receptor-ligand complex.
The Jain score is a sum of five interaction terms (Jain, 1996). These are Lipophilic interactions, Polar attractive interactions, Polar repulsive interactions, Solvation of the protein and ligand and an entropy term for the ligand. Only proximate protein-ligand atoms are considered for the pairwise interaction terms. The lipophilic and polar interaction terms are each represented by a weighted sum of a Gaussian and a sigmoidal function. This functional form is short-ranged with a pronounced maximum that occurs at close surface contacts. It also incurs a significant penalty for short contacts between protein and ligand atoms.
Parameters for Docking Study
For docking study, the Energy Grid Force Field parameter was set to Dreiding, for computing ligand-protein interaction energy. The Energy Grid parameters control the grid bases docking used in the initial evaluation of the poses. In the Dreiding force field the Gasteiger charging method is employed to calculate the partial charges of ligands and proteins. The Energy Grid Extension from site was set to 5.0 Å. The Conformation search Number of Monte Carlo Trial was set to “0” to perform a rigid docking. Maximum poses for ligand in the receptor cavity was set to 10. Ligand poses in the receptor cavity were evaluated using LigScore1, LigScore2, PLP1, PLP2, PMF, Jain, Dock Score empirical scoring functions.
Molecular properties of genaniol and limonene were analysed, to identify, if they are satisfying Lipinski rule of 5. According to Lipinski rule of 5, for any druggable compound, molecular weight should be less than 500; number of H-donors less than 5; number of H-acceptor less than 10; and octanol-water partition coefficient (ALogP) value should be less than 5. Calculated molecular properties values of geraniol and limonene are shown in Table 1. Rotatable bonds of genaniol and limonene are shown in Figure 3. Geraniol contains total 5 rotatable bonds, while limonene has only 1 rotatable bond. Ligand conformations were generated using search small molecule confirmation tools available in Accelrys discovery studio. Systematic search method was used with energy threshold 20 kcal/mol to generate total 56 conformation poses of geraniol (Table 2). Energy plot of all 56 confirmation poses of geraniol is shown in Figure 4. In limonene, only 1 rotatable bond was present, thus only 3 conformation poses were generated using systematic search conformation generation method at energy threshold 20 kcal/mol (Table 3). Energy plot of all 3 confirmation poses of limonene is shown in Figure 5. HMG-CoA reductase chain A was analysed for all the possible binding sites (Figure 6). These active sites were selected from the receptor according to their volume of the binding cavity. Docking was performed by selecting one site at a time. Binding site 2 with 326 interacting points and 40.750 Å ^3 volume showed interaction with both geraniol and limonene (Figure 7). Best poses for each geraniol and limonene with HMG-CoA reductase were analysed for different energy parameters.
| Ligand | Mol. Formula | Mol. Weight | No. of H_Acceptors | No. of H_donors | AlogP | No. of Rotatable Bonds |
|---|---|---|---|---|---|---|
 |
C10 H18 O | 154.249 | 1 | 1 | 2.934 | 5 |
 |
C10 H16 | 136.234 | 0 | 0 | 3.502 | 1 |
Table 1: Molecular properties of Geraniol and Limonene.
| Confirmation index | Angle 1 | Angle 2 | Angle 3 | Angle 4 | Angle 5 | Relative Energy | Energy |
|---|---|---|---|---|---|---|---|
| 0 | 178.238 | 288.239 | 343.105 | 304.69 | 301.204 | 7.44572 | 50.0316 |
| 1 | 179.262 | 168.06 | 342.579 | 304.651 | 301.937 | 7.96441 | 50.5503 |
| 2 | 178.916 | 286.762 | 104.3 | 305.295 | 299.859 | 1.93152 | 44.5174 |
| 3 | 178.128 | 167.4 | 104.412 | 305.207 | 299.881 | 1.60827 | 44.1941 |
| 4 | 60.6897 | 171.495 | 100.998 | 304.873 | 300.957 | 18.4569 | 61.0428 |
| 5 | 178.176 | 288.753 | 343.164 | 65.0501 | 300.378 | 5.50335 | 48.0892 |
| 6 | 179.093 | 167.801 | 342.991 | 66.3717 | 298.548 | 7.02377 | 49.6096 |
| 7 | 180.618 | 165.701 | 101.815 | 69.0623 | 299.17 | 18.9884 | 61.5743 |
| 8 | 181.456 | 44.8951 | 225.639 | 61.0061 | 303.736 | 15.073 | 57.6589 |
| 9 | 174.811 | 170.853 | 227.034 | 60.0737 | 304.949 | 15.066 | 57.6519 |
| 10 | 178.19 | 288.769 | 343.133 | 185.549 | 299.275 | 4.57314 | 47.159 |
| 11 | 179.119 | 167.91 | 342.856 | 185.862 | 298.753 | 6.00858 | 48.5944 |
| 12 | 178.899 | 286.839 | 104.229 | 185.361 | 299.33 | 0.369732 | 42.9556 |
| 13 | 295.353 | 292.389 | 98.4318 | 185.164 | 298.612 | 17.8395 | 60.4254 |
| 14 | 178.18 | 167.447 | 104.337 | 185.311 | 299.288 | 0.018356 | 42.6042 |
| 15 | 60.8582 | 171.667 | 100.883 | 184.951 | 298.309 | 16.8683 | 59.4542 |
| 16 | 182.279 | 43.9313 | 226.301 | 187.387 | 295.475 | 14.6417 | 57.2276 |
| 17 | 175.09 | 170.56 | 226.68 | 187.671 | 295.75 | 12.1075 | 54.6933 |
| 18 | 177.996 | 289.147 | 342.882 | 303.963 | 60.7323 | 17.6641 | 60.25 |
| 19 | 179.726 | 168.673 | 341.645 | 303.091 | 62.4863 | 16.1245 | 58.7103 |
| 20 | 178.675 | 287.051 | 104.57 | 303.021 | 62.2214 | 6.34724 | 48.9331 |
| 21 | 177.67 | 167.226 | 105.03 | 302.726 | 62.3295 | 5.92371 | 48.5096 |
| 22 | 178.187 | 288.763 | 343.142 | 64.0312 | 61.7222 | 4.63044 | 47.2163 |
| 23 | 179.108 | 167.848 | 342.915 | 65.0618 | 60.3784 | 6.30299 | 48.8888 |
| 24 | 177.059 | 291.348 | 101.869 | 70.372 | 44.1838 | 5.65112 | 48.237 |
| 25 | 179.766 | 166.912 | 102.457 | 69.9686 | 44.437 | 6.75359 | 49.3394 |
| 26 | 181.38 | 44.9906 | 225.574 | 60.0213 | 64.1675 | 13.5505 | 56.1364 |
| 27 | 174.744 | 170.753 | 227.108 | 58.4664 | 65.7236 | 13.7757 | 56.3616 |
| 28 | 178.19 | 288.769 | 343.133 | 184.843 | 60.5746 | 4.59975 | 47.1856 |
| 29 | 179.116 | 167.893 | 342.888 | 184.768 | 60.724 | 6.05384 | 48.6397 |
| 30 | 178.899 | 286.839 | 104.229 | 184.634 | 60.6536 | 0.340797 | 42.9266 |
| 31 | 295.359 | 292.377 | 98.4477 | 183.873 | 61.2033 | 17.8911 | 60.477 |
| 32 | 178.18 | 167.447 | 104.337 | 184.556 | 60.6654 | 0.001579 | 42.5874 |
| 33 | 60.8537 | 171.675 | 100.891 | 183.4 | 61.4292 | 16.9265 | 59.5124 |
| 34 | 182.364 | 43.8285 | 226.388 | 186.164 | 57.409 | 14.6264 | 57.2122 |
| 35 | 175.097 | 170.568 | 226.685 | 186.514 | 57.8005 | 11.7829 | 54.3688 |
| 36 | 178.17 | 288.82 | 343.087 | 304.695 | 178.643 | 6.55064 | 49.1365 |
| 37 | 179.264 | 168.071 | 342.557 | 303.874 | 178.702 | 8.04005 | 50.6259 |
| 38 | 178.916 | 286.762 | 104.3 | 304.676 | 178.553 | 2.00141 | 44.5873 |
| 39 | 178.128 | 167.4 | 104.412 | 304.564 | 178.525 | 1.67498 | 44.2608 |
| 40 | 60.6915 | 171.399 | 101.035 | 303.869 | 176.535 | 18.5049 | 61.0908 |
| 41 | 184.126 | 46.0999 | 225.24 | 287.926 | 143.951 | 14.745 | 57.3308 |
| 42 | 178.187 | 288.763 | 343.143 | 64.6461 | 181.316 | 4.5938 | 47.1796 |
| 43 | 179.106 | 167.838 | 342.936 | 65.7858 | 180.442 | 6.27443 | 48.8603 |
| 44 | 177.06 | 291.342 | 101.883 | 71.7591 | 190.121 | 6.68425 | 49.2701 |
| 45 | 178.347 | 167.413 | 104.03 | 67.0523 | 185.102 | 8.86106 | 51.4469 |
| 46 | 181.431 | 44.9197 | 225.631 | 60.595 | 183.6 | 13.7376 | 56.3234 |
| 47 | 174.747 | 170.761 | 227.115 | 59.3521 | 185.007 | 13.8229 | 56.4087 |
| 48 | 178.19 | 288.769 | 343.133 | 185.193 | 180.07 | 4.55102 | 47.1369 |
| 49 | 179.118 | 167.904 | 342.871 | 185.353 | 180.094 | 5.99049 | 48.5763 |
| 50 | 178.899 | 286.839 | 104.229 | 184.995 | 180.111 | 0.348484 | 42.9343 |
| 51 | 295.331 | 292.407 | 98.4122 | 184.587 | 180.408 | 17.7122 | 60.298 |
| 52 | 178.18 | 167.447 | 104.337 | 184.931 | 180.104 | 0 | 42.5858 |
| 53 | 60.8559 | 171.59 | 100.926 | 184.26 | 180.46 | 16.7734 | 59.3593 |
| 54 | 182.413 | 43.7613 | 226.427 | 186.802 | 176.185 | 14.7068 | 57.2927 |
| 55 | 175.099 | 170.567 | 226.675 | 187.084 | 176.559 | 11.7129 | 54.2988 |
Table 2: Confirmation poses of Geraniol generated using systematic search with energy threshold 20 kcal/mol.
| Confirmation index | Angle 1 | Relative Energy | Energy |
|---|---|---|---|
| 0 | 144.977 | 0 | 42.4817 |
| 1 | 264.12 | 5.57912 | 48.0608 |
| 2 | 24.1618 | 15.9883 | 58.47 |
Table 3: Confirmation poses of Limonene generated using systematic search with energy threshold 20 kcal/mol.

Figure 4: Energy plot of all 56 confirmation poses of Geraniol.

Figure 5: Energy plot of all 3confirmation poses of Limonene.

Figure 6: 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase (chain A). Molecular surface is colored based on calculated interpolated charges. Protein back bone is displayed as solid ribbon and colored by secondary structure type. Binding site 2 is shown with green dots.

Figure 7: Detail view of binding site in 3-hydroxy-3- methylglutaryl-CoA (HMG-CoA) reductase (chain A). All amino acids, surrounding the binding sites are labeled. Surface is colored based on calculated interpolated charges.
HMG-CoA reductase -geraniol Interaction
Docking was performed with all 56 conformation poses of geraniol and top 8 poses were analysed in the binding cavity of HMG-CoA reductase (Table 4). The best pose of geraniol (with dock score = 9.448) interacting with threonine 809, aspartic acid 767 and glycine 765 of HMG-CoA reductase. The hydrogen atom at position 29 of geraniol interacts with hydrogen atom at position 22 of threonine present at position 809 of HMG-CoA reductase. Same hydrogen atom at position 29 of geraniol interacts with oxygen of the C=O of glycine present at position 765 of the receptor molecule. Hydrogen at position 20 of geraniol interacts with hydrogen beta 1 of aspartic acid at position 767. Total ligand internal energy for the best post is calculated to 7.642. (Figure 8).
| Ligand | Conformation pose | LigScore1 | LigScore2 | -PLP1 | -PLP2 | Jain | -PMF | Dock Score |
|---|---|---|---|---|---|---|---|---|
| Geraniol | 1 | 0.86 | 2.72 | 45.43 | 46.43 | 0.46 | -7.24 | 9.448 |
| 2 | 0.7 | 2.48 | 31.06 | 32.85 | -0.28 | -4.61 | 8.023 | |
| 3 | 0.77 | 2.57 | 30.55 | 32.64 | -0.26 | -4.77 | 7.93 | |
| 4 | 0.55 | 2.47 | 44.12 | 47.07 | 1.55 | -11.37 | 5.818 | |
| 5 | 0.54 | 2.47 | 44.43 | 47.28 | 1.48 | -11.1 | 5.805 | |
| 6 | 0.55 | 2.48 | 44.48 | 47.14 | 1.56 | -10.45 | 5.786 | |
| 7 | 0.07 | 2.09 | 40.79 | 41.66 | -0.25 | -7.02 | 5.202 | |
| 8 | 0.23 | 2.29 | 51.9 | 53 | 1.05 | -4.42 | 3.424 | |
| Limonene | 1 | 0.32 | 2.6 | 26.07 | 25.32 | -0.52 | 8.56 | 15.035 |
| 2 | 0.03 | 2 | 25.88 | 28.97 | 1.67 | -7.25 | 10.593 | |
| 3 | -0.55 | 1.09 | 32.31 | 36.06 | 1.3 | -15.66 | 0.736 |
Table 4: Conformation poses of geraniol and limonene with different scoring functions. Poses are arranged with the descending dock score value.

Figure 8: Geraniol interaction with HMG-CoA reductase. All interacting amino acids with the ligand are labeled.
HMG-CoA reductase -limonene Interaction
For limonene and HMG-CoA reductase interaction, we generated three conformations using small molecule conformation generation method. All three poses of limonene were analyzed in the binding cavity of HMG-CoA reductase. The best pose of limonene has dock score as high as 10.593, much more than the best dock score in case of geraniol (Table 4). Best pose of limonene has three atoms interacting withHMG-CoA reductase. These are hydrogen at position 20, 17 and carbon at position 3. Hydrogen at position 20 and carbon at position 3 form bond with hydrogen atom of glycine amino terminus at position 808. The second interaction is in between hydrogen at position 17 of limonene and H-β1 of aspartic acid present at position 767 of HMG-CoA reductase (Figure 9).

Figure 9: Limonene interaction with HMG-CoA reductase. All interacting amino acids with the ligand are labeled.
Molecular docking studies provide lead to determine the potential of ligand interaction in the binding cavities of receptor molecules. Considering the high dock score and low ligand internal energy, it can be concluded that limonene has greater binding affinity with HMG-CoA reductase and thus having better antitumor activity in comparison to geraniol. Aspartic acid at position 767 of HMG-CoA reductase is interacting with both geraniol and limonene. This amino acid acts as a major anchor point for the ligands to interact with the receptor molecule for their anti-tumor activities.
SKG acknowledge Director, IITR, Lucknow for his generous support and CSIR NWP-17 project for providing necessary infrastructure for the work.