Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2015) Volume 4, Issue 1
More and more studies have shown that genetic determinants may mediate variability among persons in the response to a drug, and thus some therapeutics may benefit only a subset of patients. Genomic technologies, such as DNA sequencing, mRNA transcript profiling, and comparative genomic hybridization, are providing biomarkers to predict who are most likely to respond to a given drug, and thus brings opportunity to conduct targeted clinical trials with eligibility restricted to the subset of patients. In this paper, we evaluate the relative cost of a targeted design versus an untargeted design for a randomized phase III clinical trial comparing a new treatment to a control. Our investigation indicates that the effectiveness of the targeted design critically depends upon the difference of treatment effect between patient subsets, the proportion of targeted patients in the population, the diagnostic assay performance, and the relative cost of screening versus drug expenses.
<Keywords: Biomarkers, Clinical trial, Targeted design
The evidence that genetic determinants may mediate variability among persons in the response to a drug is increasing. After the intake of identical doses of a given agent, some patients may have no therapeutic response, while others may have clinically significant side effects. Caraco [1] points out that some of this diversity in response rates might be attributed to differences in the rate of drug metabolism, especially by the cytochrome P-450 superfamily of enzymes. For example, ten isoforms of cytochrome P-450 are responsible for the oxidative metabolism of most drugs. The effect of genetic polymorphisms on catalytic activity, however, is most evident for three isoforms: CYP2C9, CYP2C19, and CYP2D6. Among these three, CYP2D6 has been most extensively studied and is involved in the metabolism of about 100 drugs, including beta-blockers and antiarrhythmic, antidepressant, neuroleptic, and opioid agents. Several studies have classified some patients as having a “poor metabolism” for certain drugs due to lack of CYP2D6 activity [2,3]. On the other hands, patients having some enzyme activity are classified into three subgroups: those with “normal” activity (or extensive metabolism), those with reduced activity (or intermediate metabolism), and those with markedly enhanced activity (or ultra-rapid metabolism). Most importantly, the distribution of CYP2D6 phenotypes varies with race. For instance, the frequency of the phenotype associated with poor metabolism is 5 to 10 percent in white populations but only 1 percent in Chinese and Japanese populations.
Another example regarding the impact of genetic factors on the responses to therapeutics is the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (Iressa). Recently, Iressa was approved in Japan and the United States for the treatment of non-small cell lung cancer (NSCLC). The EGFR is a promising target anticancer therapy because it is more abundantly expressed in lung carcinoma tissue than in adjacent normal lung. Clinical trials, however, have revealed significant variability in the response to gefitinib, with higher responses observed in Japanese patients than in a predominantly European-derived population (27.5% versus 10.4%, in a multiinstitutional phase II trial) [4]. Paez et al. [5] also shows that somatic mutations of the EGFR were found in 15 of 58 unselected tumors from Japan and 1 of 61 from the United States. Treatment with Iressa causes tumor regression in some patients with NSCLC; this occurs more frequently in Japan. Finally, the striking differences in the frequency of EGFR mutation and response to Iressa between Japanese and U.S. patients raise general questions regarding variations in the molecular pathogenesis of cancer among patients.
Genomic technologies such as DNA sequencing, mRNA transcript profiling, and comparative genomic hybridization [6] are providing evidence that many diseases are more molecularly heterogeneous than previously recognized. Efforts are underway to develop mutation signatures and gene expression signatures of tumors [7,8]. Such studies provide comprehensive insights into the heterogeneity of disease pathogenesis and allow molecular disease taxonomies to be defined. Over the past decade, anticancer drug discovery has shifted from an approach based on empirical random screening to a more rational and mechanistic, target-based one [9].
Traditional drug discovery usually takes an empirical approach, characterized by random screening of a variety of natural and synthetic compounds using high throughput cell-based cytotoxicity assays. Also a traditional phase III randomized clinical trial is often conducted to evaluate a test regimen, comparing it with a control regimen for all patients in the disease category. These traditional approaches may lack efficacy, since molecularly targeted drugs may only be effective for a subset set of the patients with a traditionally defined disease. In recent years, successful target based clinical studies can be found in the literature. For example, herceptin increases the clinical benefit of first-line chemotherapy in metastatic breast cancer patients with overexpressed HER2 (the human epidermal growth factor receptor 2) level [10]. Another example is that use of Tafinlar can give a delay of tumor growth (progression free survival) by 2.4 months for unresectable or metastatic melanoma patients with BRAF V600E mutation compared with use of dacarbazine [11].
As recognized, pharmaceutical development is a risky, complex, and costly endeavor. The current paradigm for drug development was developed for the twenty century, and is no longer functioning for the 21st century. New concepts, strategies, and methodology are urgently needed to reduce the development cost, to shorten the development cost, and to improve the development success rate. The advances in genomic technologies bring the opportunity to conduct targeted clinical trials with eligibility restricted to the subset of patients predicted to have benefit from the drug. Maitournam and Simon [12] evaluate the relative efficiency of a targeted clinical trial design to an untargeted design for a randomized clinical trial comparing a new treatment to a control with regard to number of patients required for randomization. In this paper, we evaluate the relative cost of the untargeted randomized phase III clinical trial design versus the targeted design based on the required drug cost and the screening cost.
In this paper, the cost calculation of conducting a targeted clinical trial or an untargeted clinical trial is presented in Section “Cost Calculation”. We compare the cost of the two designs in Section “Comparison of Trial Cost”. Some concluding remarks are given in Section “Discussion”.
Cost Calculation
Assume that we focus on only the phase III clinical trials for comparing a test product (T) and a placebo control (C). We also assume that a binary genomic composite biomarker (diagnostic assay) can be used to classify patients potentially eligible for a particular clinical trial into two mutually exclusive genomic subgroups: those classified as marker positive (R+), who are predicted to be responsive to the new drug, and those classified as marker negative (R-), who are not predicted to be responsive. The genomic composite biomarker could be either a single biomarker, e.g. HER2 [13], or derived from genomic technologies such as DNA sequencing and mRNA transcript profiling [14,15]. In this study, a continuous efficacy response is chosen as the primary endpoint of the phase III randomized clinical trial. Let μ0 and μ1 denote the mean responses in the control group for patients in subsets R+ and R-, respectively, and let γ denote the proportion of patients in R− . For treatment T, the mean responses are denoted by μ0T and μ1T for the R- and R+ subsets, respectively. For the untargeted trial design, the response of a patient has a mixture distribution with mean γ μ0 + (1−γ )μ1 for control group C, and γμ0T+(1-γ)μ1T for treatment group T. Assume that the responses within each group and subset are normally distributed with constant variance σ2. Let n denote the number of patients per group required to reach power 1-β for rejecting the null hypothesis at significance level α, and let nt denote the number of R+ patients for the targeted design. Since the assaying of patients as belonging to subset R- or R+ is based on a genomic composite biomarker, there might exist measure error. Let λsens denote the sensitivity of the diagnostic assay for diagnosing R+ patients and let λspec denote the corresponding specificity. The mean response for patients selected for the targeted study becomes
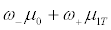
for the control group, and
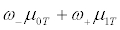
for the treatment group, where
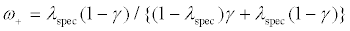 (1)
(1)
is the positive predictive value (PPV) of the diagnostic assay, and ω- = 1−ω+ . That is, the treatment effect for the targeted design is diluted by ω+. Accordingly, by Maitournam and Simon [12], we can derive that
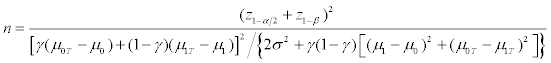 (2)
(2)
and
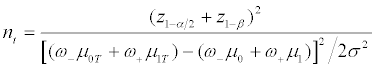 (3)
(3)
The total number of required randomized patients will be 2nt for the untargeted design, and 2nt for the targeted design. The ratio r between the required numbers of patients for the untargeted design versus the targeted design is r=n/nt. Since there exists measure error in assaying of patients as belonging to subset R- or R+, if 2nt is the number of patients required for randomization to the targeted trial, then
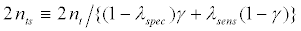 (4)
(4)
is the expected number of patients screened in order to obtain 2nt randomized patients.
Let cd denote the drug cost for a patient in the clinical trial. For the targeted design, we further need a genomic composite biomarker to classify potentially eligible patients into R+ or R- . Let cs denote the cost for screening a patient into the subsets R+ or R-. Note that the cost, cs, only affects the targeted design. Consequently, the cost for conducting an untargeted clinical trial is 2n ct whereas the cost for conducting a targeted clinical trial is
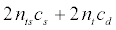 (5)
(5)
It will be worthwhile to conduct a targeted clinical trial when 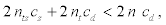
or equivalently, the ratio between the costs for the two designs, denoted by rc , is greater than 1, where
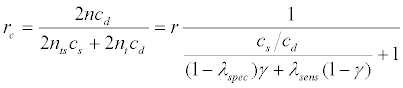 (6)
(6)
and r=n/nt is the relative efficiency of the targeted versus the untargeted design defined by Maitournam and Simon (2005). As seen in equation (6), the ratio rc depends upon r, cs /cd , λsens, λspec, and γ.
We assume that the number of patients required for randomization with each design is to achieve 80 percent statistical power at a 5 percent two-sided significance level. In other words, α=0.05 and β=0.2, and thus 1 2 z 1.96 −α = and 1 z 0.84 −β = . Here, we consider the cases with cs/cd =0.1, 0.5 and 0.9, respectively. In each case, we consider three scenarios: (I) no treatment effect exists in the R− patients, (II) the treatment effect for the R− patients is half as large as for R+ patients, and (III) the treatment effect for the R- patients is almost as large as for R+ patients, respectively. In the last scenario, we simply assume that the treatment effect for the R- patients is four-fifths as large as for the R+ patients. In comparing the targeted and the untargeted designs, we explore the following factors: the proportion of R+ patients, the relative treatment effect of the R- patients versus the R+ patients, the sensitivity of the diagnostic assay for detecting R+ patients (λsens), and the specificity of the diagnostic assay (λspec). Without loss of generality, we assume that μ0=0, and σ=1.
The relative cost, rc, of conducting an untargeted trial versus a targeted trial is shown in Figures 1, 2, and 3, which corresponds to cs/cd=0.1, 0.5, and 0.9, respectively. The horizontal axis of each plot is the proportion of R+ patients, 1- γ. In each figure, the upper panel is scenario I, and based on μ1=0 and μ1T=1. The middle panel corresponds to scenario II. The lower panel represents scenario III. The three columns of panels in each figure correspond to assay specificities of 1, 0.8, and 0.6, respectively; and the three curves in each plot correspond to assay sensitivities of 1, 0.8, and 0.6 (Figure 1).
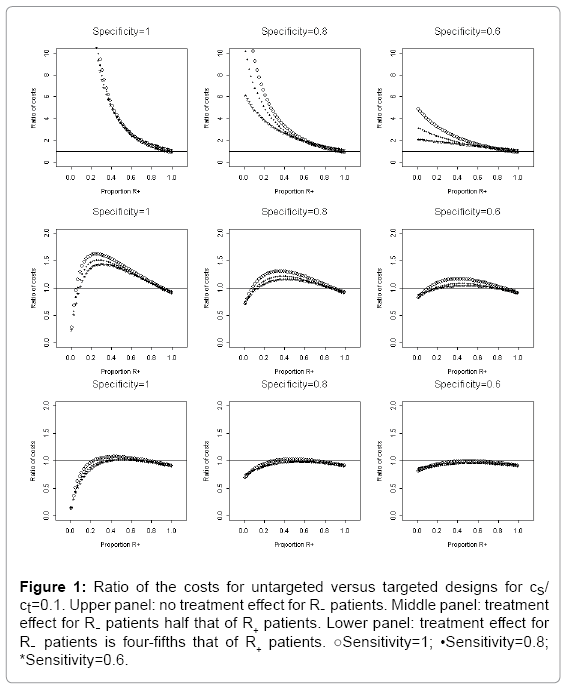
Figure 1: Ratio of the costs for untargeted versus targeted designs for cs/ ct=0.1. Upper panel: no treatment effect for R- patients. Middle panel: treatment effect for R- patients half that of R+ patients. Lower panel: treatment effect for R- patients is four-fifths that of R+ patients. ○Sensitivity=1; •Sensitivity=0.8; *Sensitivity=0.6.
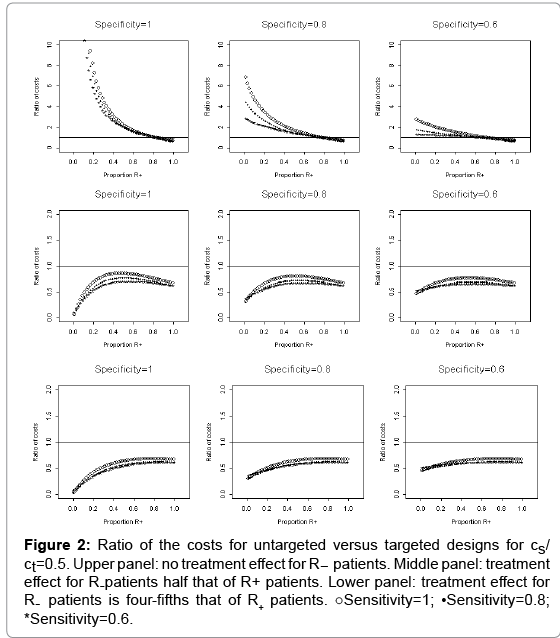
Figure 2: Ratio of the costs for untargeted versus targeted designs for cs/ ct=0.5. Upper panel: no treatment effect for R- patients. Middle panel: treatment effect for R-patients half that of R+ patients. Lower panel: treatment effect for R- patients is four-fifths that of R+ patients. ○Sensitivity=1; •Sensitivity=0.8; *Sensitivity=0.6.
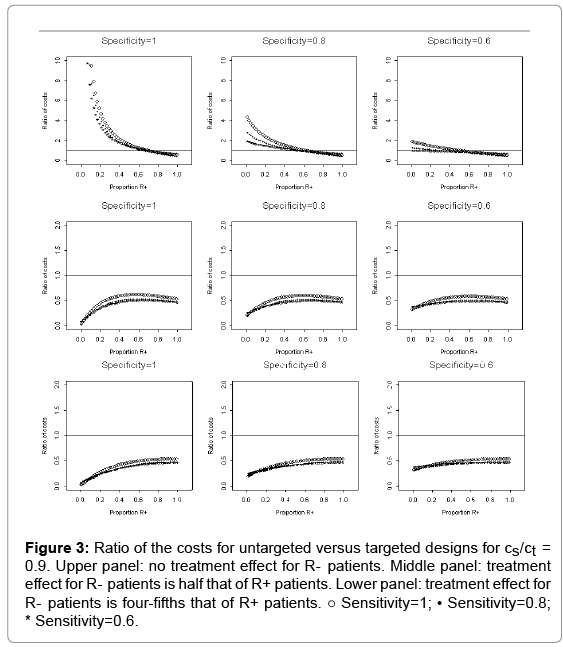
Figure 3: Ratio of the costs for untargeted versus targeted designs for cs/ct = 0.9. Upper panel: no treatment effect for R- patients. Middle panel: treatment effect for R- patients is half that of R+ patients. Lower panel: treatment effect for R- patients is four-fifths that of R+ patients. ○ Sensitivity=1; • Sensitivity=0.8; * Sensitivity=0.6.
We first evaluate the relative cost rc of the untargeted design versus the targeted design with various values of cs/cd. In Figure 1 with cs/ cd =0.1, the upper panel of Figure 1 shows that, under the scenario I (i.e. no treatment effect for R- patients), the cost for conducting an untargeted trial is much higher than that for conducting a targeted trial (i.e. ratio rc is greater than 1) even in the worst situation when both the sensitivity and the specificity are 0.6. When the treatment effect for the R- patients is half as large as for R+ patients (the middle panel) and the assay specificity is 1, conducting the targeted trial is cheaper than the untargeted trial (the relative cost rc is greater than 1), except for the extreme cases where the proportion of patients in R+ is close to 0 or close to 1. As the assay specificity decreases to 0.6, the relative cost rc decrease to almost 1; hence, there is no significant benefit in cost to conduct a targeted trial. When the treatment effect for the R− patients is four-fifths as large as for R+ patients (cf. the lower panel), the relative cost rc is close to 1 or less than 1. Therefore, it is less worthwhile to conduct a targeted trial. Note that the curves corresponding to different assay specificity have different shapes. Therefore, the specificity plays a more important role than the sensitivity does with regard to rc. If the specificity decreases, rc decreases because some R- patients selected for inclusion in the targeted trial will dilute the treatment.
For cs/cd =0.5, Figure 2 shows that the cost for conducting an untargeted trial is lower (i.e. rc is less than 1) than that for conducting a targeted trial when the R- patients have treatment effect (cf. the middle and lower panels). This suggests that there is no substantial advantage to conduct a targeted trial when the treatment effect for R- patients is half that of the R+ patients or the treatment effect for R- patients is as large as the treatment effect for R+ patients. However, when there is no treatment effect for the R- patients, it may be worthwhile to conduct a targeted trial since ratio rc is greater than 1, especially when the assay specificity is 1 (cf. the first plot of the upper panel of Figure 2). The relative cost rc, however, is not much larger than 1 when the assay specificity decreases to 0.6 (cf. the last plot of the upper panel of Figure 2). Thus, there is no significant benefit to conduct a target trial when the assay specificity λspec=0.6 given cs/cd =0.5 (Figure 2).
When cs/cd =0.9, Figure 3 shows that it is not worthwhile to conduct a targeted trial since the ratio rc is much less than 1 in both scenarios II and III (i.e., there is some treatment effect for the R- patients). The cost for conducting an untargeted trial is higher (i.e. the ratio rc is greater than 1) than that for conducting a targeted trial only in the best situations when the assay specificity is 1, there is no treatment effect for the R- patients, and the proportion of the R+ patients is less than 0.6. To sum up, it can be seen from Figures 1, 2, and 3, that as the ratio of screen cost per person and drug cost per person, cs/cd , increases, the savings in trial cost for conducting the targeted trial will decrease significantly (Figure 3).
We now turn to analyze the trend of the relative cost of conducting an untargeted trial versus a targeted trial given different assumption for the treatment effect in R- patients. If γ is close to 0, then most of the patients are in the R+ group. Thus the two trial designs are almost equivalent and the cost for targeted design is higher than that for untargeted design since the former spending the additional cost of screening for each patient. We conclude that rc is less than 1 when γ is close to 0 for each scenario. We now consider the scenario (I), i.e., no treatment effect exists in the R- patients. In this case, we can assume μ1 = μ0 = μ0T . When γ is close to 1, there are very few R+ patients. The treatment effect for the untargeted trial design can be expressed as
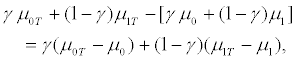
which is close to zero. Hence, the untargeted trial requires a huge sample size based on equation (2). In this scenario, if the treatment effect in the R+ subset is small, then the sample size required for the targeted design will also become very large based on equation (3). That is, both designs require large numbers of patients for the trial. Consequently, conducting a targeted trial will have no advantage. On the other hand, if the treatment effect for R+ patients is large, the targeted design may require very few patients. However, the untargeted design may still require a large number of patients due to no treatment effect for the R- patients. Accordingly, the cost of the untargeted design is very high due to the large number of required patients. In this case, if the screening cost cs is much lower than the drug cost cd, then the cost of the targeted design will be smaller than that of the untargeted design. In other words, the relative cost rc is much larger than 1 (cf. upper panel in Figures 1, 2 and 3). In this case, conducting a targeted trial is cheaper than conducting an untargeted trial.
In the scenario II, where the treatment effect for the R- patients is half as large as for R+ patients, the net treatment effect for the targeted design is
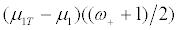
For γ close to 1, there are very few R+ patients, and the treatment effect for the untargeted trial design is again
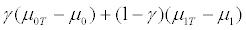
which is close to 0 0 ( T ) μ −μ , and the treatment effect for the targeted trial design is
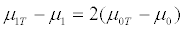
which is two times larger than that for the untargeted trial design. Hence the untargeted trial requires a sample size approximately twice that of the targeted trial. When both γ and λspec are close to 1, by equation (4), the expected number of patients screened in order to obtain 2nt randomized patients is enormous. Hence, the total cost of screening 2nts patients could be huge when the screening cost per patient, cs, is not very small relative to the drug cost per patient, cd, in the clinical trial. This implies that the cost of the untargeted trial could be much lower than that of the targeted design when the ratio cs/cd is not very small. Accordingly, the relative cost, rc, of the untargeted design versus the targeted design is less than 1, and thus conducting the targeted trial should have advantage over the untargeted trial.
In the scenario III, where the treatment effect for the R- patients is four-fifths as large as for R+ patients, the net treatment effect for the targeted design is
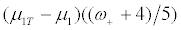
Again, when γ is close to 1, there are very few R+ patients and the treatment effect for the untargeted trial design is
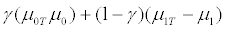
On the other hand, the treatment effect for the targeted trial design is
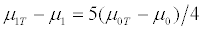
which is slightly larger than that for the untargeted trial design. Hence the untargeted trial requires a slightly larger sample size than the targeted trial. When both γ and λspec are close to 1, the expected number of patients screened in order to obtain 2nt randomized patients is huge by equation (4). Thus, the cost of conducting an untargeted trial could be lower than that of conducting a targeted trial since the total cost of screening 2nts patients could be huge. Therefore, the relative cost of the untargeted design and the targeted design, rc, could be less than 1. That is, conducting a targeted trial is more expensive than conducting an untargeted trial.
Clinical trials are evolving. The current paradigm for drug development was developed for the twenty century, and is no longer functioning for the 21st century. Therefore, new concepts, strategies, and methodology are urgently needed to reduce the development cost, to shorten the development cost, and to improve the development success rate. In this paper, we have compared the untargeted randomized phase III clinical trial design to the targeted design based on the required drug cost and the screening cost for both designs. When the treatment effect exists only for R+ patients and the specificity of the diagnostic assay is close to 1, untargeted designs would generally cost more than targeted designs. As the specificity of the assay decreases, the cost of the targeted design increases but the cost of the targeted design remains lower than that of the untargeted design. If, however, the screening tool sensitivity is poor (e.g., 0.6), then the cost for the targeted design will increase dramatically.
When the treatment effect for the R- patients is half that of the R+ patients or as large as for the R+ patients, it can be seen that the number of patients to be screened for the target design is usually larger than the number of patients required for the untargeted design. Consequently, the cost for the targeted design would be much greater than that for the untargeted design unless the screening cost is very cheap. It should also be noted that the increased number of screened patients for the targeted design strongly depends on the sensitivity of the diagnostic assay. That is, the larger the sensitivity of the assay, the less the total screening costs. Thus, the sensitivity of the screening tool has critical impact on the cost for the targeted design.
Our investigation has also indicated that the specificity of the diagnostic assay and the treatment effect for the R- patients play important roles on the relative costs of the untargeted design versus the targeted design. More specifically, the larger the specificity, the larger the relative costs of the untargeted design versus the targeted design. On the other hand, the larger the treatment effects for the R-patients, the smaller the relative costs of the untargeted design versus the targeted design. In other words, the targeted design remains advantageous with regard to the cost issue when the diagnostic assay specificity is large, or when the treatment effect for the R- patients is not expected. The selection of the preferred design will also depend on the cost of the diagnostic assay. In particular, the advantage of the targeted design increases, as the relative cost of screening versus drug expense decreases.
In this study, our investigation was only focused on the cost of the diagnostic assay and the drug expenses. However, there will be more time needed for planning and development, and more money spent on clinic staff, lab expenses, and trial management cost for targeted designs. This additional cost may be included in the evaluation of the overall cost of the clinical development.