Journal of Chemical Engineering & Process Technology
Open Access
ISSN: 2157-7048
ISSN: 2157-7048
Research Article - (2023)Volume 14, Issue 3
Enzymes are biochemical catalysts that facilitate chemical reactions under Physiological conditions. Currently enzymes are being employed in industrial biotechnology for numerous purposes for the production of novel and sustainable products at a speedy rate. Enzyme technology is the change of an enzyme's structure or catalytic activity in order to produce new metabolites or participate in new reaction pathways. Simultaneously, significant technical advancements are encouraging the chemical and pharmaceutical sectors to embrace enzyme technology, a movement fueled by worries about health, energy, raw resources, and the environment. The therapeutic and financial success of small-molecule enzyme inhibitors, such as kinase inhibitors in oncology, enzyme targets are a key focus of contemporary drug research and development activities. Understanding the progression of an enzyme-catalyzed reaction can aid in conceptualizing different types of inhibitors and informing the design of screens to uncover desired pathways. Similarly, much of the current drug discovery and development work is focused on identifying and developing therapeutic candidates that act by inhibiting specific enzyme targets. The high levels of illness association and drag ability that characterize this family of proteins make enzymes appealing as drug development targets. The current practices and future directions in drug discovery enzymology in this expert opinion, with an emphasis on how a detailed understanding of the catalytic mechanism of specific enzyme can be used to identify and optimize small-molecule compounds that interact with conformationally distinct forms of the enzyme, resulting in high potency, high selectivity inhibitors. This review highlights the classical concepts of Enzyme technology and opens new routes for drug discovery.
Enzyme inhibitors; Enzyme substrate; Slow-binding; Tight-binding; Drug
People now have a longevity and health-care quality that is unsurpassed in the history of the world. Much of the improvement in life quality and duration, particularly in the developed world, may be ascribed to better sanitation (including the availability of drinkable water), better diet, and the availability of modern health care. The creation and distribution of pharmacological substances to treat many of the world's ailments has made a substantial contribution to better human health. Orally-dosed medications that may be easily supplied to patients for easy and simple self-dosing outside of critical care facilities are very beneficial in this regard. As a result, orally accessible, small-molecule medicines have been and continue to be a fundamental basis of modern medicine as well as the pharmaceutical business. Several attempts have been made in recent years to categorise the sorts of biologic macromolecules that serve as targets for present pharmacological treatments [1].
Enzymes are physiological catalysts that play key roles in a variety of biological processes, such as cell development and division, cellular signalling, and metabolism. Because of the presence of distinct substrate-binding pockets, which may be used as binding sites for pharmacological enzyme inhibitors, they are appealing therapeutic targets [2]. Enzyme inhibitors and inactivators account for about half of all marketed medications, and they have revolutionised human medicine. Angiotensin-Converting Enzyme (ACE) inhibitors, such as captopril and lysinopril, which first appeared in the late 1970s, are today considered to be the most significant family of medications for the treatment of hypertension. The rational design of ACE inhibitors was based on the Mechanism of Action (MoA) of this metalloproteinase [3]. Furthermore, statins, such as atorvastatin and lovastatin, developed in the 1980s as natural product-derived inhibitors of 3-hydroxy-3-glutaryl CoA reductase and are today the most often used medications to treat hypercholesterolaemia [4]. Beginning in the 1990s, saquinivir and indinavir were discovered to be rationally designed inhibitors of HIV-1's aspartic protease. HIV-1 protease inhibitors have significantly aided in the transformation of HIV/AIDS from a 'death sentence' to a treatable condition. Furthermore, ATP-competitive inhibitors of protein kinases have given an arsenal of anticancer and anti-inflammatory medications during the last 20 years [5].
Unsurprisingly, enzymes now account for one-third or more of the distinct therapeutic targets identified in leading pharmaceutical firms' portfolios. This is true even for pharmaceutical businesses that have shifted their focus to biologic medicines. Because biopharmaceutical medicines are often limited to external drug targets, intracellular enzymes may still be a promising field for drug development [6]. Understanding the role of enzymes in disease states and developing techniques to modify their activity for therapeutic benefit remains a major focus of drug research. While enzymology is a powerful and well-established science, it is sometimes overlooked in terms of offering scientific insight into challenges encountered along the path from target discovery to clinical proof of concept. This is especially true at the start of an enzyme-targeted drug discovery strategy. It is unusual to have a comprehensive understanding of an enzyme target's kinetic mechanism (the sequence of substrate addition and product release) and chemical mechanism of catalysis before the start of 'hit' discovery.
Of course, enzymology alone cannot provide all of the necessary data or information in early drug discovery, and for a comprehensive pharmacological picture of the thermodynamics and kinetics of enzyme catalysis and inhibition, its conjunction with biophysical techniques and protein structure studies is still necessary. Furthermore, integrating knowledge gained from detailed mechanistic characterization in isolated enzyme assays with cell-based assays and, eventually, metabolic pathways and systems knowledge will maintain enzymology as one of the most important quantitative disciplines in drug discovery.
This article will highlight the significance of doing high-quality mechanistic enzymology investigations, consider how thorough and early information of an enzyme mechanism may add value throughout a drug-discovery process, and evaluate the worth of integrating such knowledge with other data.
Enzyme substrates
Another crucial factor is the identity, or biochemical character, of the enzyme substrates. There are several instances when binding interactions between macromolecular substrates and enzymes take place away from the active site, and these interactions might in fact add to the overall binding energy required to form the first Enzyme-Substrate (ES) encounter complex. In particular, if specific conformational changes occur in conjunction with native substrate binding, substitution of the native substrate with, for example, a shorter peptide in the case of a protease or kinase could affect both the ability to identify inhibitors that might prevent formation of the ES complex by interacting with distal sites and the kinetic mechanism of the enzyme. For instance, in bacterial Glu-tRNA Gln aminotransferases, distinct inhibitor pockets are exposed following substrate binding [7]. There are several instances of protein kinases where the substrate choice directly affects the kinetic mechanism and small-molecule affinity. The findings indicate a rapid-equilibrium, random-order process of substrate addition (or ordered with ATF2 protein binding first) when MAPK p38α catalyses phosphoryl transfer to the cAMP-dependent transcription factor ATF2. However, the reaction follows an ordered pathway with MgATP binding first when a short peptide acts as the phospho-acceptor. For the 3-Phophoinositide-Dependent protein Kinase 1 (PDK1), phosphorylation of an extended peptide substrate containing a single distal recognition motif replies via a rapid equilibrium, random-order mechanism, it is known that a similar situation exists [8]. However, a steady-state ordered process in which S6K1 binding prevents MgATP association leads to the phosphorylation of the natural downstream substrate S6K1 protein kinase. Control over the particular downstream pathways that can be suppressed in a biological environment can be achieved by a thorough understanding of the catalytic mechanism and careful study of the numerous enzyme-substrate interactions. For instance, differential substrate selectivity in the case of p38 can pinpoint the precise pathways that small molecules influence, providing precision control and possibly fewer undesirable side effects [9].
The need to use native nucleosome substrates rather than short histone N-terminal tail peptides as surrogates has been recognized in the field of epigenetics for a number of years in order to find small-molecule inhibitors that translate into the cellular context and target the desired activity of the enzyme. Prioritizing particular processes of interest to target is made possible by having in-depth information of the molecular environment of the enzyme in the context of the illness. A new focus in the field of kinase drug discovery is on the identification of inhibitors that are either non-competitive inhibitors or uncompetitive inhibitors with respect to ATP, in order to get around the problem of high cellular concentrations of ATP. For instance, it may be preferable to identify inhibitors that are not competitive with the native substrate where the cellular concentration of the latter is high. Although uncommon, enzyme inhibitors that work through uncompetitive inhibition are suitable for stymieing the activity of enzyme targets inside metabolic pathways because the build-up of the substrate that results from their usage cannot be competitively used to overcome the inhibition.
Enzyme kinetics: Most enzyme-catalyzed processes have rate limits imposed by chemical or product release stages. Under steady-state circumstances in the latter scenario, an enzyme-product complex will predominate, especially at high substrate concentrations. An enzyme-product complex or a mimic of it might be enhanced in the test conditions in a chemical screening technique. By scanning an enzyme species with a partly blocked active site in this manner, HTS may find a 'hit' molecule that would inhibit the enzyme by blocking the desorption of the product ligand. The target enzyme would typically be inhibited by such an inhibitor in an uncompetitive manner. The development of an inhibited ternary complex may give more selectivity than merely binding to the free enzyme, and screening an enzyme-product complex samples a different inhibitor space than screening the free enzyme. In such an HTS campaign, a single plate may be screened twice: once without the addition of a product analogue and once with one. The first plate would show competitive or mixed-type inhibitors, but the second plate would show uncompetitive inhibitors more frequently. For instance, the release of the product NAD+ is the rate-limiting step of the bacterial enoyl reductases encoded by the FABI and INHA genes. Hit compounds that bind more tightly to the enzyme-nicotinamide complex rather than the free enzyme may provide a more desirable series of hit compounds for lead optimization if one were to conduct HTS in the presence and absence of added NAD+ or its analogue, such as acetyl-pyridine adenine dinucleotide [10].
Designing inhibitors that demonstrate kinetic selectivity, in which the drug-target complex has a longer half-life than off-target-drug complexes, may also need careful consideration of the rates of binding and desorption, k(on) and k(off). Many medications that are now on the market have slow-binding inhibitory kinetics, and the idea of residence time has proven crucial in translating these kinetics into cellular and in vivo contexts. In an animal model of infection, a prospective mechanistic pharmacodynamics model that includes drug-target kinetic parameters, such as the on and off rates for the formation and breakdown of the drug-target complex, has recently been described. This model is intended to predict dose-response curves for inhibitors of the LpxC enzyme from Pseudomonas aeruginosa [11]. Also gaining popularity is irreversible covalent inhibition, which achieves the required target selectivity without sacrificing safety or raising concerns about off-target toxicity by adjusting the ratio between the initial molecular recognition event and chemical reactivity [12].
Enzyme inhibitor: A chemical that binds to an enzyme and inhibits its activity is known as an enzyme inhibitor. Proteins called enzymes speed up the chemical processes that turn substrate molecules into products and are essential for life. By attaching the substrate to its active site, a unique region on the enzyme that expedites the process's most challenging stage, an enzyme speeds up a particular chemical reaction [13].
An enzyme inhibitor halts (or "inhibits") this process by attaching to the enzyme's active site and preventing the substrate from binding there or by attaching to another place on the enzyme and preventing the enzyme from catalysing the reaction (Figures 1A and 1B). Inhibitors of enzymes can bind irreversibly or reversibly. Irreversible inhibitors join forces with the enzyme to establish a chemical connection that prevents the enzyme from working until the bond is severed. Reversible inhibitors, on the other hand, bind non-covalently and have the potential to spontaneously depart from the enzyme, allowing it to resume its normal activity. Reversible inhibitors can either bind to the enzyme, the enzyme-substrate complex, or both, which results in various forms of inhibition.

Figure 1: A: Enzyme (E) at speeds up the process of turning substrates (S) into products (P); B: By attaching to the enzyme, the inhibitor (I) prevents the substrate from attaching. Binding site is depicted as a black rectangle, a black checkerboard, and a rounded green rectangle.
Since they typically only block one specific enzyme and work to regulate the activity of that enzyme, enzyme inhibitors are essential components of all cells. For instance, compounds generated later in a metabolic route may block enzymes in the process, reducing the synthesis of molecules that are no longer required. A cell must have this kind of negative feedback in order to remain balanced. Additionally, enzyme inhibitors regulate necessary enzymes like nucleases and proteases that, if unregulated, may harm a cell. Enzyme inhibitors are toxins created by animals or plants that prevent the action of vital enzymes in prey or predators.
Enzyme inhibitor as drug: Many medication compounds are enzyme inhibitors that block a pathological pathogen, such as a virus, bacteria, or parasite, or an abnormal human enzyme. Examples include protease inhibitors for treating HIV/AIDS and methotrexate, a drug used in chemotherapy and rheumatoid arthritis. Anti-pathogen inhibitors often target just one enzyme, making them extremely selective and, assuming no human analogues for that enzyme exist, generally having little adverse effects in humans. Given the genetic distance between these diseases and humans, this is frequently the case. Since the dissociation constants of pharmaceutical enzyme inhibitors.
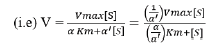
Here α and α' represents the inhibitor concentration and its two dissociation constants
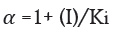
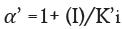
are frequently low, very little of the inhibitor is needed to block the enzyme. The risk of liver and kidney damage as well as other negative medication responses is decreased in humans when the enzyme inhibitor is present in low concentrations. Consequently, the development of enzyme inhibitors is a current topic of biochemistry and pharmacology study.
Enzyme inhibitors are most frequently used as pharmaceuticals to treat illness. Many of these inhibitors attempt to treat a pathological condition and target a human enzyme. For instance, the commonly used medicine aspirin inhibits the cyclooxygenase enzyme's ability to commit suicide. As a result of this inhibition, proinflammatory prostaglandin synthesis is suppressed, and aspirin can be used to treat pain, fever, and inflammation [14].
Enzyme inhibitors, of which about one-fifth are kinase inhibitors, make up an estimated 29% of authorised medications as of 2017. The receptor tyrosine kinases, which are crucial enzymes that control cell development and whose over-activation may lead to cancer, are a prominent class of kinase therapeutic targets. As a result, kinase inhibitors like imatinib are widely employed to treat cancer [15]. Another well-known example of a drug's target enzyme is janus kinases. A number of inflammatory disorders, including as arthritis, asthma, and Crohn's disease, are treated with Janus kinase inhibitors because they prevent the release of inflammatory cytokines.
Sildenafil (Viagra) (Figure 2) is a frequent medication for erectile dysfunction. This substance has a strong inhibitory effect on the enzyme cyclic guanosine monophosphate specific phosphodiesterase type 5, which breaks down the signalling molecule. This signalling molecule stimulates smooth muscle relaxation and opens the corpus cavernosum to blood flow, which results in an erection. The medicine lengthens the duration of the signal by reducing the activity of the enzyme that stops the signal [16].

Figure 2: The sildenafil (Viagra) molecule.
Slow and tight binding inhibitors: Enzyme inhibitors can work either irreversibly or reversibly [17,18]. There is a continuum in the time-dependent characteristics of enzyme inhibitors, and certain substances that covalently inactivate an enzyme exhibit greater reversibility than a noncovalent inhibitor that forms a stunningly tight binary complex. Both in vitro and in vivo, these inhibition mechanisms exhibit highly dissimilar behavior. Reversibility, however, is not an absolute phenomenon since inhibition that is thought to be irreversible may really be reversible over a period that is far longer than the typical assay duration. This results in an operational definition that categorizes inhibition as irreversible if the loss of enzyme activity brought on by an inhibitor is not reversible over the timescale of the enzyme activity assay [19,20].
Since there is no chemical reaction involved in reversible inhibition, merely a straightforward noncovalent contact, full inhibition is typically attained quickly. The development of covalent bonds between a reversible inhibitor and the enzyme is possible, even though irreversible inhibitors are not necessary to do so. Only kinetic factors are used to distinguish between irreversible and reversible inhibitors. Slow-binding inhibitors are substances that lie in the range of reversible and irreversible inhibition [21-26]. Slow-binding inhibition, which typically precedes tight-binding inhibition, is a common occurrence during drug discovery [6]. This nomenclature, however, may be misleading because many of these substances bind to the enzyme quickly before slowly isomerizing into a form with higher affinity. For instance, this is the mechanism behind the inhibition of INHA [26-34] by the isoniazid-NAD adduct, as well as some metal-binding inhibitors of HIV-1 integrase [35]. The inhibitor's dissociation happens slowly as well. Since off rates are usually slow for a slow-binding inhibitor, on rates can be quick or sluggish. Operationally, slow binding is characterized as an increase in inhibition during the experiment. Irreversible inhibition might be viewed as a particular kind of sluggish binding in which the enzyme activity recovers too slowly to be noticed.
Slow binding inhibition: A single IC50 number may not be a valid indicator of inhibitory efficacy because the degree of inhibition for a slow-binding inhibitor rises over time, causing the IC50 to vary as well. Instead of using single time points for dose-response analysis, it is critical to understand the significance of analyzing time courses for inhibitors suspected of being slow binding or irreversible. Failing to do so could lead to inaccurate annotation of potency and misleading information regarding the MoA. The rate constants of the processes that result in the formation of the enzyme-inhibitor (EI) complexes must instead be characterised. For instance, slow association and dissociation of Vertex-11e and SCH772984 were observed in the presence of extracellular signal-regulated kinase 1 (ERK1; also known as MAPK3 and ERK2) and ERK2, with apparent kon values of 0.21 and 2.8 M-1 s-1 and dissociation rate constants of 0.21 and 1.1 h-1, respectively [36]. The majority of slow-binding interactions are accounted for by two significant, separate mechanisms [20].
The first happens because of the law of mass action, which states that reactions will move more slowly at low concentrations. The values of the pseudo-first-order rate constants for the formation of the non-covalent enzyme-inhibitor (EI) complex (k3 (I)t) and the dissociation rate constant (k4) would be low when an inhibitor has a low inhibition constant (Ki) and the inhibitor concentration (I) is varied in the region of Ki. Low k3 values can also lead to slow-binding inhibition. Even though k3 may be on the order anticipated for a diffusion-controlled reaction (>108 M-1 s-1), these low values of association and dissociation would result in delayed binding. It can also be shown that if (E) is very low, potentially because most of (E)t is actually in the form ES due to the presence of a very highly competitive substrate, delayed binding may follow. The genuine second-order rate constant (k), (I), and the enzyme concentration (E) are all factors in this equation. The quick creation of an initial collision complex (EI), which then goes through gradual isomerization to become a higher affinity complex (EI*), can also result in slow-binding inhibition. It has been proposed that mechanism B accounts for the majority of slow-binding and slow, tight-binding inhibitions [21]. Initially joining with enzymes at their active sites, inhibitors are thought to then promote conformational changes that improve affinities and lead to the creation of a more stable EI complex, from which inhibitor is gradually released.
As a result, Ki* must be lower than Ki for inhibition to follow mechanism B, and as a result, k6 must be lower than k5. Additionally, the magnitudes of k5 and k6 must be such that it is possible to see the achievement of equilibrium between EI and EI*.
It is possible to distinguish between these two different mechanisms of slow-binding inhibition using nonlinear regression analysis (Figure 3) with the help of secondary plots of kobs versus (I), which are linear for the one-step mechanism and hyperbolic for the two-step mechanism, and to estimate the individual rate constants by following the progress curves for inhibition in reactions starting with enzyme.

Figure 3: Slow binding inhibition.
Jump-dilution experiments, where the pre-equilibrated E-I complex is rapidly diluted into the assay and recovery of enzyme activity is monitored as the inhibitor dissociates from the complex [22], are another helpful strategy. High substrate concentrations are used in these experiments, which may help to improve the observable signal by competing with the inhibitor for rebinding to the free enzyme.
Tight-binding inhibition: By its very nature, the drug development process frequently leads to the discovery of high-affinity compounds. Due to their great affinity, these substances are frequently challenging to describe. Understanding that genuine Ki values must be compared and that, in some circumstances, such as when ligand depletion occurs, IC50 numbers do not accurately represent affinities is crucial for effectively ranking drugs [13]. Though it actually refers to a condition where the active enzyme concentration is about equal to or even higher than the apparent inhibition constant, this circumstance is frequently referred to as "tight binding." This suggests that at high enzyme concentrations, strong binding could happen even for weaker molecules. This phenomenon has been known for many years through the relationship among IC50, KiÃÂ?¹ (the apparent Ki) and (E)t described by Goldsteinin [16] in 1944 (equation 1):
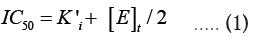
Ki is equal to 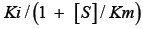 Equation 1 makes it clear that as the apparent Ki value decreases, the measured IC50 is only able to approach the apparent Ki value when the functional enzyme concentration is low compared to the apparent Ki value. Conversely, the measured IC50 can only reach half of the functional enzyme concentration. The impact on drug development is significant because as the affinity approaches the enzyme concentration, the IC50 cannot be utilized to rank compounds. Additional strategies must be used in this situation to completely comprehend the true affinity. Even data-fitting methods, such as the quadratic equation outlined by Morrison [21] in equation 2, which do not assume that the free concentration of the inhibitor equals the total concentration, become unreliable as the affinity increases past about 10-fold below the functional enzyme concentration.
Equation 1 makes it clear that as the apparent Ki value decreases, the measured IC50 is only able to approach the apparent Ki value when the functional enzyme concentration is low compared to the apparent Ki value. Conversely, the measured IC50 can only reach half of the functional enzyme concentration. The impact on drug development is significant because as the affinity approaches the enzyme concentration, the IC50 cannot be utilized to rank compounds. Additional strategies must be used in this situation to completely comprehend the true affinity. Even data-fitting methods, such as the quadratic equation outlined by Morrison [21] in equation 2, which do not assume that the free concentration of the inhibitor equals the total concentration, become unreliable as the affinity increases past about 10-fold below the functional enzyme concentration.
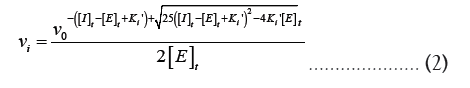
Where vi is the inhibited rate, vi is the uninhibited rate and [I]t is the total inhibitor concentration. When [I]t = IC50,
vi = 0.5 0 v , and equation 2 can be rearranged to provide equation 1.
Assays are initially configured with low concentrations of enzyme to avoid this issue; therefore, further decreases can cause issues with the biosignal sensitivity. Experimental approaches to lower the functional enzyme concentration or raise the apparent Ki by the use of increased concentrations of competing substrate may have limited utility. Only substances with a competitive component to binding can modify Ki, and frequently only small shifts may be achieved due to low solubility constraining the achievable concentration: Km ratio. Following an in silico analysis of experimental error and assay design, experimental conditions for precise determination of Ki values for tight-binding enzyme inhibitors have been described [37]. In practice, this can entail attempting to connect the dissociation constant (Kd) to Ki and/or using biophysical and/or kinetic-based approaches to quantify the constant (Figure 4).

Figure 4: Functional Enzymes concentration vs. Degree of inhibition graph. 
Mechanisms of inhibitions
In terms of how substrates or other ligands affect the degree of inhibition, test compounds may employ a variety of different mechanisms [38]. Competitive inhibition is widespread because inhibitors are frequently made to resemble an enzyme reaction's substrates or to bind at the place where the substrate binds, frequently leading to the discovery of competitive kinetics. Other processes, however, are also conceivable. One of them is non- competitive kinetics, in which a test substance may bind both before and after substrate binding (and in which the inhibition is referred to as mixed inhibition when the affinities for the free enzyme and ES complex are dissimilar). Uncompetitive inhibition, which is rarer than competitive inhibition, happens when the inhibitor binds only after the substrate has attached to the enzyme, as is the case with inosine 5'-monophosphate dehydrogenase that is covalently complexed to its nucleotide substrate (Figures 5A and 5B) [17,24,39].

Figure 5: Tight binding inhibition. Note: The inhibitor concentration is represented on a linear scale in the 5A, whereas the 5B displays the concentration-response behaviour on a semi log scale.
In order to determine the mechanism of inhibition and to extrapolate the activity seen in isolated protein assays to effects seen in cells, where the concentration of substrates may differ from that in the biochemical enzyme assay, it is crucial to understand how the concentration of substrates affects the measured IC50 values after inhibitors have been identified during primary screening. Additionally, it enables accurate selectivity assessment because the mechanism of inhibition and the substrate concentration at which selectivity is measured can affect the degree and direction of inhibitor selectivity. Several classic enzymology texts provide thorough descriptions of these observations [18]. Initial studies to ascertain the mechanism frequently concentrate on two substrate concentrations, above and below the Km of the variable substrate. It is advised to conduct comprehensive matrix-based experiments covering a variety of substrate and inhibitor concentrations rather than using this sparse data approach so that accurate Ki values may be determined. This may provide a hint of the mechanism of inhibition.
The Cheng-Prusoff equation must be modified appropriately to account for the nature of the assay system [40] and to investigate instances of cooperative binding, which may necessitate more complicated equations when the slope is not equal to 1.
Impact of enzymes
Traditional enzyme markets: Enzyme technology is particularly suited for the food, feed, agriculture, paper, leather, and textile industries since both finished goods and raw materials are made up of biomolecules that can be created, broken down, or altered by enzymatic processes. Numerous industrial applications for the many enzymes that are commercially available have been identified [27]. The editor of Food Technology recently penned an informative summary of current advancements in the food industry [29]. Enzyme sales in the United States and Europe reached roughly $22.5 billion in 2022 due to the increased usage of enzymes in the pulp and paper sectors for cleaner production processes. Technology advances are highly encouraged by the availability of clean, low-cost enzymatic or biological alternatives to chemical processes. The market value of biotechnology for clean production in the paper sector as a result ($31-62 billion) is therefore extremely substantial [28]. The use of laccases for bleaching or catalase to eliminate hydrogen peroxide reduces the need for raw materials and waste production significantly. Enzymes play a similar role in the production of textiles [27].
Enzyme technology in chemistry and pharma sectors: Due to the well-established chemical manufacturing methods and the relatively limited overall economic advantage of biotechnological processes at the moment, the industrial chemicals sector is difficult for enzyme technology to enter [28]. New biocatalytic processes need to be significantly improved, integrated with chemical and downstream processes, and capable of operating at high speeds at high substrate and product concentrations in order to be accepted. Nevertheless, the manufacture of fine chemicals and pharmaceuticals already heavily relies on enzyme-based procedures [30]. It is simpler for biotechnology to compete with conventional organic chemical processes since these product groups frequently contain novel and/or chiral chemicals. This is especially true if a new approach is more cost-effective and makes better use of raw materials and other resources. Chiral biomolecules are the targets of pharmacological substances, such as enzymes or cell-surface receptors. It follows that it is not surprising that only one enantiomer of a chiral medication has the required biological activity whereas other isomers may have no biological activity at all or an undesirable activity. Most manufacturers will utilize chiral compounds more frequently as a result of regulatory obstacles, the necessity to extend patents, and the requirement to increase the safety and efficiency of their products.
Single enantiomer medicine sales have already surpassed $100 billion [31] globally, and over half of all new drugs expected to hit the market in the next five years are expected to have optically pure active components.
The vast number of chemicals that must be evaluated for biological activity in order to identify a single potential lead is a significant problem in the pharmaceutical industry. Combinatorial biocatalysis has drawn a lot of attention in this context because it has the potential to increase the complexity of the diversity of current chemical libraries or to be utilized to create libraries from scratch [32]. One illustration is the modification of the glycosylation pattern of bioactive substances using glycosyltransferases [33].
Today, enzyme technology is only used to create a small number of common compounds, such as acrylamide (annual manufacturing scale: 40,000 tons). However, this achievement has shown that bioconversion technology may be expanded.
Dow and Cargill have begun building a large-scale polylactic acid production facility with a capacity of 40,000 tons annually; DuPont's biobased 1,3-propanediol synthesis appears to be getting near to actual production as well. On a multi-ton scale [30], biocatalysis also produces a wide range of additional molecules, such as chiral compounds [31]. For instance, Lonza in Switzerland now generates 3000 tons of nicotinamide annually from 3-cyanopyridine. According to Maxygen, biological techniques already have access to $50 billion of the $1.6 trillion chemicals business. Biotech-based goods may eventually control up to 50% of the polymer market and 15% of the market for fundamental chemicals.
Since its originations more than a century ago, molecular enzymology has advanced significantly. Recombinant expression technology has made it possible to get pure target enzymes in huge quantities, allowing for the full study of mechanistic features in both the absence and presence of inhibitors. The knowledge of disease processes and their modulation has been greatly impacted by the coupling of these mechanistic investigations with the expanding availability of high-resolution individual protein and complex structures, as well as biophysical approaches that can help probe specific enzyme forms. It is not unexpected that current drug discovery has advanced significantly during the same period of time, given that the enzymologist's sharp ideas, concepts, and methodological rigor have been applied to the scientific issues involved in drug discovery. Early prioritization of these studies makes it easier to identify undesirable mechanisms, allowing for the specific reprioritization of compounds that would otherwise cause development to be delayed due to problems like a lack of true target engagement or action via mechanisms that are not appropriate for progression. The end result of this mechanism-based, reasoned decision-making process is higher-quality drugs with a higher likelihood of being successful in clinical trials, improved regulatory submission quality, and higher-quality publications.
Due to more affordable manufacturing techniques, novel enzymes, and new application areas, the enzyme market and the number of competitive enzyme-based processes are expanding quickly. It is possible to utilize enzymes that are especially suited to their application and process circumstances thanks to the capability of directed evolution and gene shuffles, as well as effective methods to search the environment for new enzymes. Due to a number of factors, including straightforward cost savings, the rapidly rising demand for chiral chemicals, the trend towards sustainable industrial development (less waste, less CO2), and last but not least, the opportunities presented by emerging technologies, we anticipate that enzyme technology is on the verge of a significant breakthrough.
[Crossref][Google Scholar] [PubMed]
[Crossref] [Google Scholar][PubMed]
Citation: Keerthi P, Lathif AK, Nesaghi A (2023) Enzyme Technology for Drug Discovery. J Chem Eng Process Technol. 14: 471
Received: 31-Jul-2023, Manuscript No. JCEPT-23-25900; Editor assigned: 02-Aug-2023, Pre QC No. JCEPT-23-25900 (PQ); Reviewed: 17-Aug-2023, QC No. JCEPT-23-25900; Revised: 23-Aug-2023, Manuscript No. JCEPT-23-25900 (R); Published: 31-Aug-2023 , DOI: 10.35248/2157-7048.23.14.471
Copyright: © 2023 Keerthi P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.