Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2016) Volume 5, Issue 2
Biologic drugs, as Infliximab (IFX), are widely used in inflammatory bowel disease (IBD). IFX is a chimeric monoclonal immunoglobulin directed against TNF-α and acting in IBD through several mechanisms of action. We divide these mechanisms into central and peripheral. Central mechanisms are considered effective at systemic level, like the reduction of plasmatic pro-inflammatory cytokines (TNF-α, IFN-γ), the promotion of apoptosis of inflammatory cells, the induction of T regulatory cells (FOXP3CD4+) and the modulation of cellular inflammatory pathways. Peripheral mechanisms could be considered acting at mucosal levels like normalization of endothelial functions, blockade of angiogenesis, restoring balance between matrix metalloproteinases (MMPs) and tissue inhibitors of matalloproteases (TIMPs). This classification could serve as solid guide to better categorize mechanisms of lack of response to IFX, which account for up to 30% of patients per year. Central mechanism of loss of response proposed are the formation of antiinfliximab antibodies (ATI), trough serum levels, metabolic factors and pharmacogenomics; peripheral mechanisms of loss of response could be the degradation of IFX by MMPs at mucosal level and the loss of IFX trough inflamed mucosa. In the present review, we analyze mechanisms of action and loss of response to IFX in course of IBD known until today.
<Keywords: Anti-TNF α; Inflammatory bowel disease; Infliximab; Loss of response; Mucosal healing; Pharmacodynamics; Pharmacokinetics
TNF: Tumor Necrosis Factor; CD: Crohn Disease; UC: Ulcerative Colitis; IFX: Infliximab; ATI: Anti-Infliximab Antibodies; IBD: Inflammatory Bowel Disease.
Infliximab, the first anti-TNF-α agent for IBD, is a chimeric monoclonal antibody directed against tumor necrosis factor-α (TNF-α). This antibody is composed by combining a variable region of murine origin (25% of the total molecule) and a constant region of human origin (75% of the total molecule), joined through disulfide bonds. Infliximab binds with high affinity both soluble form and transmembrane form of TNF-α. Tumor necrosis factor-α is a proinflammatory cytokine that plays a key role in the pathogenesis of inflammatory bowel disease (IBD) [1]. TNF-α appears to be abundantly expressed in the tracts of intestine that are affected by inflammation, both as soluble and transmembrane form.
Transmembrane form of TNF-α, which is the precursor of the soluble form, is expressed on the surface of activated macrophages, lymphocytes and other cells. After being processed by the TNF-α conversion enzyme (TACE), soluble form of TNF-α is cleaved from trans-membrane form and thus may mediate its biological effects by binding to the receptors for TNF-α type 1 and type 2 (TNF-R1 and TNF-R2 ) (Figure 1) [2]. In this manner, TNF-α contributes to mucosal inflammation through different mechanisms including the destruction of the intestinal barrier, the induction of apoptosis of epithelial cells and the secretion of cytokines. “Biologics” in IBD are “anti-TNF-α”, and interact directly with TNF-α, thus blocking its effects [3].
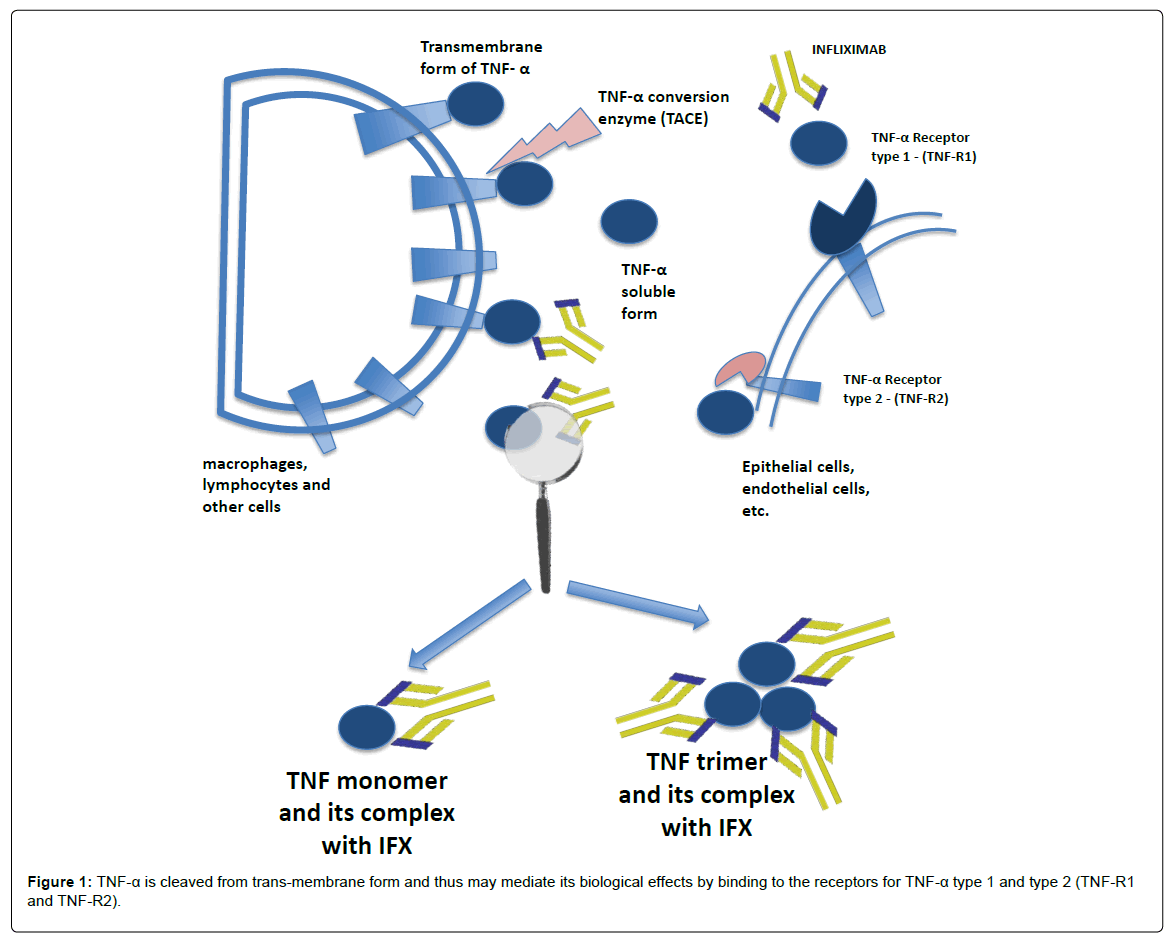
Figure 1: TNF-α is cleaved from trans-membrane form and thus may mediate its biological effects by binding to the receptors for TNF-α type 1 and type 2 (TNF-R1 and TNF-R2).
Infliximab is administered by intravenous infusion and it is indicated for the induction and maintenance of remission of moderate-severe IBD. Currently available data show that there is still a high percentage of IBD patients do not respond to Infliximab. The loss of response could be depend on several factors and mechanisms, most of them not yet completely known [1,3,4]. Many aspects of pharmacodynamics and pharmacokinetic of Infliximab are yet to be clarified. This review aims to describe mechanisms of action and loss of response to Infliximab in course of IBD known until today, with particular attention to new and emerging pathways.
Pharmacokinetics
The pharmacokinetics of infliximab is not well defined in IBD, but in many studies it is evident a linear relationship between the dose administered and plasma concentration measured.
The majority of data available in IBD deals with the so called “through levels”, which could be defined as levels of IFX just before the following infusion. These “trough or basal levels” of IFX range from 5.5 μg/ml to 14 μg/ml, and they are dose-frequency dependent. The median peak serum reported for a dose of 5 mg/kg ranges from 127 μg/ ml to 153 μg/ml; the median central volume of distribution has been calculated in 2.7 Litre which is similar to plasma volume, that suggests an unicompartimental kinetics. The estimated median serum clearance is 3.6 ml/day/kg
The half-life of IFX is longer than other biological agents directed against TNF-α and varies between 7.7 and 9.5 days. Because of the long half-life and slow elimination rate, IFX remains determinable in the serum for 8 weeks after a single infusion of 5 mg/kg [1,3,4].
In patients receiving 5 mg/kg according to the standard protocol that involves infusion at 0, 2, 6 weeks, and every 8 weeks after the third infusion, the serum concentration of infliximab is greater in the induction phase (defined as the first 14 weeks of therapy) compared to the next maintenance phase in which the serum concentration of the drug tends to stabilize over time [1,3,4].
The clearance from the systemic circulation of the anti-TNF-α has not yet been completely understood, however it seems that a mechanism of elimination can be represented by proteolysis following endocytosis mediated by a receptor expressed on cells of the reticuleendothelial system. The process of phagocytosis through which phagocytes internalize antibodies is driven by the Fc receptor-γ and is more effective if there is cross-linking between the immunoglobulin. Another mechanism of clearance is degradation in lysosomes after binding of circulating antibodies with antigens of membrane cell [5]. The clearance of the monoclonal antibodies can also be influenced by the neonatal Fc receptor (FcRn), which are expressed both by cells of the reticule-endothelial and by vascular endothelial cells that bind and internalize antibody (Figure 2) [6]. The administration of infliximab leads to highly variable concentrations among patient, explained by the variability of pharmacokinetic properties. Many studies about infliximab pharmacokinetics model identify different covariates: body weight, serum albumin concentration, immune response status and concurrent immune-modulator usage. The impact of total body weight influences infliximab clearance and central volume of distribution (per kilogram of body weight), that suggests an inadequate correction of body size by the per-kilogram dosing scheme. The decrease of central volume of distribution and peripheral volume of distribution (per kilogram of body weight) as patient total weight increases is likely due to the relatively lower vascularization with increased body fat that may be associated with increased body weight. High serum albumin concentration values are associated with low infliximab clearance, and low serum albumin concentration is associated with high infliximab clearance. Albumin, endogenous immunoglobulin’s, and monoclonal antibodies such as infliximab are protected from catabolism by the Fc receptor (FcRn). A high serum albumin concentration may indicate increased functional efficiency of FcRn, leading to efficient antibody conservation and consequently reduced monoclonal antibodies elimination. In ATI positive patients, infliximab clearance increases. Concurrent immune-modulator use is associated with a decrease in infliximab clearance [7]. Other studies show an elimination half-life shorter in the presence of ATI. These induced antibodies are more frequent when infliximab is not administered regularly. Also sex is found to significantly influence central volume of distribution: this parameter is higher in men than in women. The influence of sex may occur because, for a given body weight, plasma volume is lower in women than in men [8].
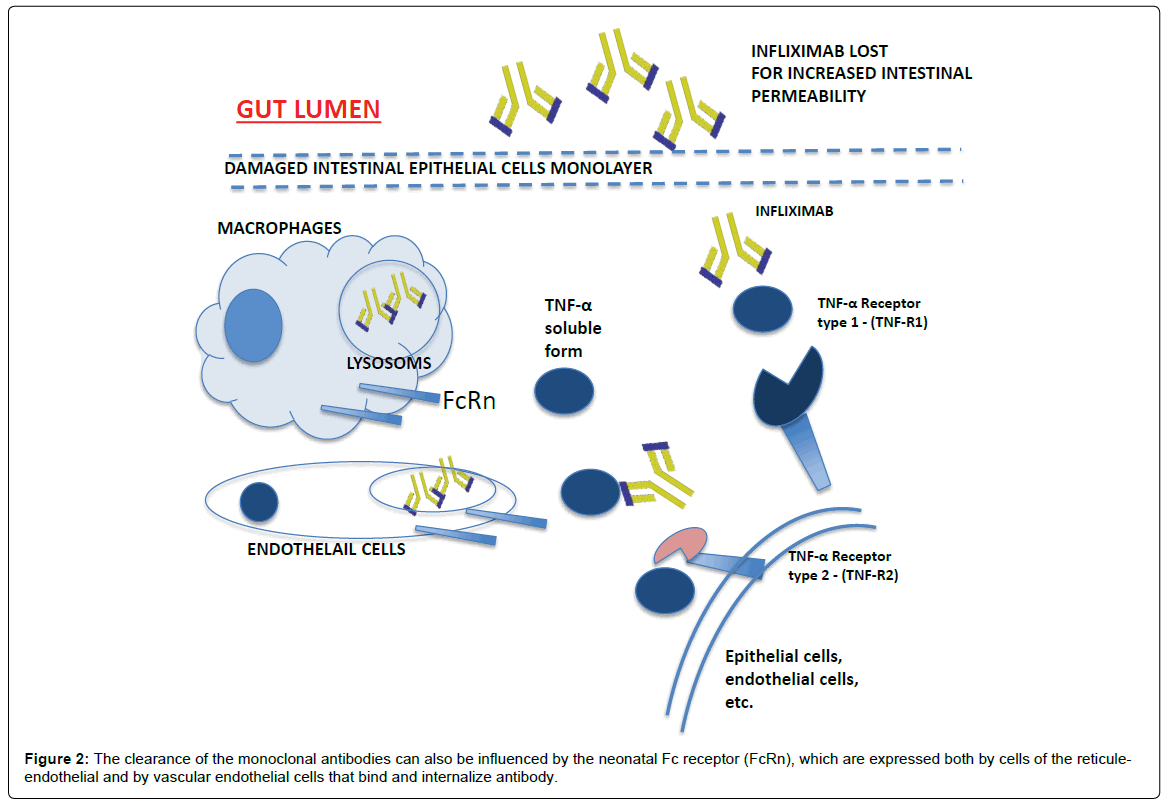
Figure 2: The clearance of the monoclonal antibodies can also be influenced by the neonatal Fc receptor (FcRn), which are expressed both by cells of the reticuleendothelial and by vascular endothelial cells that bind and internalize antibody.
Over recent years, it has been repeatedly tried to find a correlation between serum levels and clinical response to Infliximab. These levels are correlated to presence or absence of concomitant immunosuppressive therapy, and other factors related to methodology of experiment such as the timing of measurement, the chosen cut-off and the presence or absence of primary response to Infliximab [1].
In CD, serum concentration of infliximab is directly correlated to the clinical effectiveness. In fact, for values greater than 12 μg/ml a median duration of response of 81.5 days has been described, while for values less than 12 μg/ml an average duration of response 68.5 days was reported. Patients receiving concomitant immunosuppressant also more easily reach concentrations greater than 12 μg/ml. Other studies also show an inverse relationship between the concentration of infliximab and C-reactive protein [9,10].
In the ulcerative colitis (UC) patients with moderate-to-severe disease activity, it has been shown that serum concentration of infliximab, after a regimen of induction and maintenance median of 13 months, is related to an improvement of the disease in clinical and endoscopic findings; furthermore indeterminable levels of infliximab represent a risk factor for colectomy [11].
Many studies suggest that development of immunogenicity in course of therapy with anti-TNF-α agents has a direct role on the effectiveness of these drugs. A prospective study has shown that 61% of patients develop ATI after the fifth infusion, however, the incidence of ATI is much lower in patients receiving concurrent immunosuppressive drug, being observed in 43% of patients. In the same study, the ATI’s concentration was related to the duration of response which was 35 days for concentrations greater than 8 μg/ml and 71 days for concentrations below that range [9].
In another study it was shown that administration of hydrocortisone 200 mg as a premedication before infliximab, is associated to a lower incidence of ATI development in patients compared to placebo (21% vs. 29% at week 8 and 26% vs. 42% at week 16) [12].
The presence of ATI also has an influence on the safety of the drug; in fact elevated levels of this immunoglobulin are associated with a higher incidence of adverse reactions during infusions.
In conclusion, high serum levels of infliximab are related to wider duration of response and a greater window of clinical and endoscopic remission. The development of antibodies directed against the anti- TNF is associated with reduced efficacy and safety of the drug.
Mechanisms of Action of Infliximab
Infliximab forms a stable complex with the soluble or membrane TNF-α, expressed on cells within the intestinal mucosa like immune cells, fibroblasts, endothelial or epithelial cells (Table 1) [13].
| Mechanisms of action of Infliximab: |
|---|
| Central mechanisms: |
| reduction of pro-inflammatory cytokines |
| modulation of immune cell function |
| modulation of inflammatory pathways |
| Peripheral mechanisms: |
| down-regulation of epithelial apoptosis |
| restoring endothelial function |
| blockade angiogenesis |
| restoring MMPs and TIMPs balance |
| promoting mucosal healing |
Table 1: Mechanisms of action of Infliximab.
Infliximab binds to both monomer and trimer forms of soluble TNF. The monomeric form (17 kDa) of TNF is not active but its blockage possibly prevents the formation of bioactive trimer TNF. Moreover, infliximab makes complexes with TNF with great stability and slow disassociation rate, potently inhibiting its interaction with TNF surface receptors.
In addition to soluble TNF, Infliximab binds with high avidity to transmembrane TNF, forming similar stable complexes with slow disassociation. Each Infliximab molecule has the ability to bind with two soluble TNF molecules and up to three infliximab molecules can bind to each TNF homotrimer, resulting in full blockage of all receptor binding sites on TNF [14].
To better understand mechanism of actions of IFX, here we divide, mechanisms of action into central and peripheral mechanisms. Central mechanisms are considered effective at systemic level, like the reduction of plasmatic pro-inflammatory cytokines (TNF-α, IFN-γ), the promotion of apoptosis of inflammatory cells, the induction of T regulatory cells (FOXP3CD4+), the formation of ATI, trough serum levels, metabolic factors and pharmacogenomics. Peripheral mechanisms could be considered acting at mucosal levels like normalization of endothelial functions, blockade of angiogenesis, restoring balance between matrix metalloproteinases (MMPs) and tissue inhibitors of matalloproteases (TIMPs). This classification could be considered as solid guide to better categorize mechanisms of lack of response to IFX.
Central Mechanisms of Action
Modulation of circulating immune cell function and vitality
Infliximab induces apoptosis in immune cells while binding to transmembrane TNF. This effect has been verified on in vitro studies, showing induction of apoptosis in peripheral T cells and intestinal lamina propria mononuclear cells from IBD patients, or also on CD8+ cells [14].
Furthermore, infliximab binding transmembrane-TNF-transfected Jurkat T cells induced interleukin-10 production, G0/G1 cell cycle arrest, accumulation of reactive oxygen species and up-regulation of Bax, Bak, ROS, IL-10, and p21WAF1/CIP1 [15]. Infliximab treatment was associated also to an increase in peripheral blood regulatory T cells FOXP3CD4 [16,17]. Regulatory T cells play an important role in the pathogenesis of IBD and a change in Tregs count might be a direct effect of IFX, while an increase in these cells may act as a biomarker for the response to anti-TNF therapy [18]. Finally IFX is associated to the modulation of T cell immunoglobulin- and mucin-domain-containing molecule 3 (TIM-3), human β-defensin (HBD)-2, forkhead box protein 3 (FOXP3), and the frequency of CD4+ CD25+ FOXP3+ regulatory T cells (Tregs) in children with Crohn’s disease (CD) [19].
Modulation of inflammatory pathways
Infliximab promotes the activity and expression of mitogenactivated protein kinases (MAPKs). These kinases play a crucial role in the organization of the inflammatory response. In mammalian cells have been identified four subgroups of MAPKs: ERKs, JNKs, p38 kinase and ERK5/big MAPK. In vivo studies show a significant increase of p38 in the first 48 hours of infusion of the drug [20]. In vitro studies on peritoneal macrophages isolated from TNF-R1 knockout mice shows that IFX failed to counteract MAPK activation, I-kB degradation, phosphorylation of Ser536 on the NF-kB subunit p65 and iNOS and COX-2 expression. On the other side, it is also showed a protection in a concentration-dependent manner on activation of TNF-R1 wild type macrophages stimulated with LPS/IFN [21].
Finally, in Rheumatoid arthritis patients, proteins of the p38 signalling network were proposed as prognostic factors to IFX response [22].
Modulation of intestinal mucosa immune cell function and vitality
CD patients treated with IFN infusion showed an increased number of CD3+ T cells in apoptosis at TUNEL assay performed on colonic biopsies [23]. Infliximab is able to promote T-cells apoptosis in IBD-affected mucosa through the binding to transmembrane TNF, complement activation, and/or triggering of antibody-dependent cell-mediated cytotoxicity. Because transmembrane TNF is also expressed by macrophages, Infliximab treatment could promote macrophage apoptosis through a mechanism that involves binding of transmembrane TNF and triggering of macrophage/NK cell–mediated cellular cytotoxicity or complement-mediated lysis.
Pro-inflammatory cytokines reduction
In IBD, a highly abundance of TNF by immune cells has been described, although not uniformly, both in peripheral blood and within intestinal mucosa [24,25]. Infliximab has been shown to primarily reduce concentration of mucosal TNF, which in turn reduces other cytokines, like IFN-γ [26]. In another paper, IFX has been associated to the reduction of the mucosal growth factor macrophage (GM-CSF) [27].
Very recently regulatory macrophages have been described as novel regulators of innate immunity precessess: it seems that anti-TNF antibody induce them through a membrane receptor Fcin order to produce anti-inflammatory cytokines and express CD206 [28].
Down-regulation of epithelial apoptosis
The loss of integrity of the intestinal barrier, caused by apoptosis of enterocytes, is an important step in the pathogenesis of IBD. The mainly responsible of apoptosis of enterocytes is TNF-α. Consequently, infliximab interfering with this pathway reduces apoptosis by decreasing the loss of integrity of the intestinal barrier [16].
Restoring endothelial function and blockade angiogenesis
In IBD, expression of endothelial leukocyte adhesion molecules, as ICAM and VCAM, is increased; consequently there is a greater recruitment of immune cells within intestinal mucosa. Following infusion of IFX, the normal expression of leukocyte adhesion molecules is restored. Furthermore in IBD patients, IFX modulated the responsiveness of endothelial cells to acetylcholine, another important mechanism regulating vasodilation and flow of leukocytes [29]. An in vitro study showed that infliximab treatment significantly reduced levels of plasma sCD40L and eliminated CD40 and VCAM-1 from mucosal micro-vessels in treated patients with CD [30]. Finally IFX regulates angiogenesis. Recently it was reported that following IFX infusion, both endothelial proliferation marker Ki67 and mucosal concentration of VEGF were reduced [31].
Modulation of inflammatory pathways
The balance between matrix metalloproteinases (MMPs) and tissue inhibitors of matalloproteases (TIMPs) is essential for the homeostasis of the intestine. The metalloproteinaase ADAM 17, also known as TACE (TNF-α converting enzyme), is essential for the conversion of the membrane form of TNF-α in the soluble form and its concentration within the intestinal mucosa appears to be increased in IBD patients. Recently it has been shown that also other MMPs, including ADAM 19 have a higher concentration compared to healthy controls in the intestinal mucosa of these patients. Infliximab causes an increase in the production of TIMP-1 by restoring the balance between MMPs and TIMPs [32-34]. In a recent paper it was observed that in patients with active IBD, gene expression of many MMPs, TIMPs, ADAMTSs, and growth factors were upregulated, while colonic expression of MMP28 and TGFα were reduced. Infliximab therapy was associated to a complete restoration of these genes in responders. Furthermore increased ratio of MMP1/TIMP1 expression at baseline in active IBD was restored in responders with colonic mucosal healing [35,36]. The nuclear transcription factor kb (NF-kB) is one of key factor in IBD pathogenesis. Infliximab treatment was associated to an increased concentration of NF-kB inhibitors, like IkBα and IkBγ, with a consequent lower secretion of TNF at the level of the mucosa [37].
Promoting mucosal healing
The mechanism through which infliximab improving mucosal healing could be represented by the induction of regulatory macrophages (CD206+). This was suggested in a paper in which it was shown that this induction of regulatory macrophages is present in patients with mucosal healing but is absent in those who did not show signs of mucosal healing [28,38]. To support the hypothesis of a direct role of infliximab in mucosal healing, our group recently shown that infliximab reduces infiltration of macrophages (CD68+) and lymphocytes (CD3+) in the inflamed mucosa of IBD patients, and at the same time it is observed an increasing expression of E-cadherin and fibroblast growth factor (FGF). These data correspond to a restoration of glandular structures and epithelial cells of the mucosa [39].
Another study showed that the main mechanisms associated with the resolution of endoscopic inflammation in patients with IBD treated with infliximab are the reduction of CD68+ macrophages and the decreased expression of IL-17A. Indeed, downregulation of the expression of macrophage-associated genes highly correlated with the endoscopic response to infliximab treatment. Significant level of correlation was also found between the decreased expression of IL-17A gene and the endoscopic responses in patients with IBD. Both Cd68 and IL-17 were downregulated at higher level in patients achieving mucosal healing (good responders) compared to patients not achieving mucosal healing (intermediate/low responders). This study also demonstrates that infliximab downregulates the mucosal expression of IL-23 and Nos2a (gene encoding the inducible nitric oxide synthase enzyme), together with several antigen-presenting cell costimulatory molecules, raising the possibility that a reduced antigen-presenting cell–mediated stimulation of Th17 cells could account for the diminished IL-17A expression observed in our cohort of patients [40].
Primary and secondary lack of response to infliximab (IFX) is the major challenge in treating IBD. The ACCENT I trial showed already a lack of response in about half of patients. Mechanisms are still not fully elucidated; however different patterns have been proposed [41-43].
In these paragraphs we will focus on mechanisms of loss of response to IFX. These can be divided into “central mechanisms” and “peripheral mechanisms”, according to the site where the loss of response occurs.
Anti-infliximab antibodies (ATI)
The most critical issue in loss of response seems to be the immunogenicity of the molecules, leading to production of antiinfliximab antibodies (ATI). ATI may contribute to clinical loss of response both indirectly by compromising bioavailability through local formation of immune complexes (non-neutralizing ATI) and directly by preventing the interaction between the drug and TNF-α (neutralizing ATI). Based on these findings, regular monitoring of both circulating drug levels and neutralizing and non-neutralizing ATI, appears the best way to tailor anti-TNF-α therapies. However, in many cases the presence of ATI is associated with a greater elimination of the drug [44,45].
Approximately 60% of patients exposed to IFX develop ATI, responsible also for acute reactions during the infusion and loss of response to treatment. However, ATI are not determinable in 25% of patients who lose response to IFX. This implies the presence of other mechanisms involved in this process (Table 2) [46].
| Loss of response mechanisms of Infliximab: |
|---|
| Central mechanisms: |
| anti-Infliximab antibodies (ATI) |
| Other autoantibodies (ANA and dsDNA) |
| Low trough serum concentration |
| drugs and metabolic factors |
| Peripheral mechanisms: |
| shift of disease targets |
| pharmacogenomics and pharmacokinetics |
| Infliximab degradation by MMPs and mucosal cells |
| consumption/loss of infliximab trough inflamed mucosa |
Table 2: Loss of response mechanisms of Infliximab.
In a proportion of patients (28%) with ATI, ATI can disappear and the response to IFX can be restored. The disappearance of ATI after the beginning of immunosuppressive therapy was observed in 20% of patients with transient ATI. In 20% of patients ATI disappear after dose optimization, however no clear reasons have been identified in the majority of cases (60%) [47].
Factors that would influence the formation of anti-drug antibodies include drug dose, dosing intervals, induction regimen followed by a scheduled therapy rather than an episodic therapy (on demand), concomitant use of immune-modulators, prophylactic steroids before infusion. Data from clinical trials and prospective cohort studies demonstrate that episodic administration of IFX is associated with higher rates of ATI than scheduled maintenance therapy (up to 60% compared with <5%) [48].
Anti-drug antibodies may be directed against non-human molecular structures, as is the case of IFX, in which the main immunogenic component is represented by the murine part of the Fab fragment. Anti-drug antibodies are less frequently detected when humanized molecules are used [49].
Others autoantibodies
In a prospective study the role of antinuclear antibodies (ANA) and anti-double-stranded DNA (Anti-dsDNA) was investigated in patients with psoriasis not responding to IFX. Non-responders patients displayed higher levels of these autoantibodies and concentrations of these antibodies was even higher in patients exposed to different anti-TNF drugs [50]. These are the first reported data to suggest that the development of ANA and anti-dsDNA antibodies on anti-TNF treatment is a marker of forthcoming treatment failure in patients with psoriasis and indeed any patient groups being treated with anti-TNF agents. This is probably due to the apoptotic effect of the drug and to the resulting exposure of nuclear fragments to the immune system in presence of low levels of C-Reactive Protein (CRP), which is a scavenger of nuclear fragments. The low levels of CRP can be attributed to anti- TNF drugs, which decrease the CRP serum concentration [10].
Another study shows that patients exposed to IFX for at least one year, develop anti-dsDNA and anti-ssDNA, being more prevalent in patients who have also developed ATI [51].
The exact role of ANA in the loss of response of IFX, hovewer, is not still not completely defined and large-scale prospective studies are required to investigate whether their development is associated with anti-TNF treatment failure.
Trough serum concentration
Circulating drug levels can be considered a marker of TNF-α neutralizing capacity and a potential useful kit to adjust active IFX treatment. The finding of high drug trough levels in primary nonresponder patients, in fact, should discourage optimization of IFX therapy and open the orizons to newer therapeutic targets [49]
On the other side, undetectable serum levels of IFX are associated with loss of response of patients with active IBD and this may occurs also in patients not developing ATI. Clinical remission has been clearly associated to detectable trough serum levels of the drug. On the other hand, it has also been demonostrated that serum IFX levels increase together with an increase in clinical response when IFX dosage is increased by shortening intervals of administration or increasing dosage of it from 5-10 mg/kg [52].
A prospective study in patients with Crohn's disease shows that patients with IFX trough levels <2.5 μg/mL (measured 4 weeks after the first infusion) have a positive predictive value (86%) for development of high ATI (>8 μg/mL). This finding is similar to what it has been described for arthritis, where lower doses and concentrations of IFX are associated with higher rates of ATI during treatment initiation.
The immunogenicity of lower doses of IFX was also reduced by concomitant methotrexate in patients receiving lower doses of IFX. Thus, the FDA-approved-doses of IFX, used in practice for IBD (5 mg/ kg), may be less immunogenic when concomitant immune-modulators are used. It is unclear if post-hoc addition of an immune-modulator would allow a return to lower doses of IFX in patients who need dose escalation [53].
Drugs
One of the first studies assessing interaction of IFX with other drugs comes from a cohort of patients with rheumatoid arthritis, showing that IFX elimination is reduced in patients under active treatment with methotrexate [54]. Other studies showed that IBD patients in active treatment with azathioprine and IFX displayed lower IFX serum levels following the discontinuation of the immune-suppressant [55].
Nutritional status and albumin
A recent study showed that also nutrional status, and in particular albumin, could potently enter into the IFX homeostatis. In particular, albumin levels have been suggested to be predictors of serum concentration of the drug and clinical response. Patients with high levels of albumin maintain higher serum concentration of IFX, lower clearance and a longer half-life than patients with low levels of albumin; the mechanism remains unknown [56].
Metabolic and environmental factors
The role of smoking, which is a notorious risk factor for Crohn's disease, has been particularly studied among environmental factors. It is associated to a reduced efficacy and to a greater loss of response to anti-TNF, being related to number of daily cigarettes [57].
A complex alteration in immune cell function in smokers has been described. The effects of smoking include induction of the inflammatory response, immune suppression, alteration of cytokine balance, and DNA damage. Studies in healthy volunteers and patients with rheumatoid arthritis show that smokers have increased TNF-α release from peripheral blood monocytes and stimulated T lymphocytes. An increase of TNF-α could lead to consumption of IFX and to reduce serum drug levels [58].
Recently, some studies assessed also the role of obesity in the loss of response to IFX. An increasing BMI (body mass index) is associated with an attenuated response and with an earlier loss of response to IFX. These observations can be explained in part by the pro-inflammatory state and the increase of cytokines that characterize adipose tissue in obese people [59].
Comorbidity
Non-IBD related inflammation, like infections, ischemic colitis, vasculitis, could be significant comorbidity in IBD, sometimes inducing a loss of response to IFX. Inflammation can increase TNF-α production then saturating drug levels, or it can activate inflammatory pathways different from TNF-α. Non-inflammatory mechanisms of comorbidities, on the other side, depends more on the type of the conditions lile a diagnosis of irritable bowel syndrome, fibrostenotic strictures, cancer, dietary (lactose ingestion, etc.), miscellaneous (amyloidosis, bacterial overgrowth, Bile salt diarrhea, etc.) [60].
Shift on disease target and pharmacogenomics
Some interesting works demonstrate that the target of the disease may change during biological therapy, as suggested by the increase in the local concentration of other pro-inflammatory cytokines, such as GM-CSF. However other authors describe a reduction of these cytokines following administration of IFX [27,61].
Other studies identified molecular predictors of response or of nonresponse, suggesting a clear importance of mucosal pathways directly or indirectly associated to TNF able to have an impact on IBD. It is the case of five genes differentially expressed in non-responders ulcerative colitis patients, like osteoproteogerin, stanniocalcin-1, prostaglandinendoperoxide synthase-2, receptor α2 of IL13 and IL 11, or TNFAIP6, S100A8, IL-11, G0S2 and S100A9 in Crohn’s disease [62,63].
Finally other studies show that IFX non-responders Crohn's disease patients have significantly higher serum CRP, sIL-2R and ANA as compared to responders group, indicating dysregulated T lymphocyte activation in non-responders. Additionally, patients with loss of response time greater than 1.5 years showed higher sIL-2R levels compared to patients with the loss of response time lower than 1.5 years, suggesting that patients with longer loss of response time had significant T cell-driven disease.
Infliximab degradation by MMPs
In inflamed intestinal mucosa there is an increased expression of MMPs, particularly MMP-3 and MMP-12. Same researchers showed, with preliminary data, that the levels of these MMPs decrease after infusion of IFX in patients responding to the treatment; on the contrary, high levels of MMP-3 and MMP-12 are observed in patients not responding to IFX [64]. Biancheri and colleagues presented preliminary data which show that some MMPs, especially MMP-10 and MMP-12, have a direct role in the degradation of anti-TNF-α agents, in particular MMP- 12 [65].
Consumption/loss of infliximab trough inflamed mucosa
Recently it has been shown that detectable levels of IFX can be find within intestinal mucosa and stools. In particular, animal affected by DSS colitis displayed lower IFX levels within serum, intestinal mucosa together with an higher level of IFX on fecal samples, compared to control non-colitic animals. This observation suggests that inflammation may lead to an increased consumption of the drug together with an increased loss of the drug through inflamed mucosa within feces [66]. Similarly, results have been presented by our group in human samples at several conferences as preliminary results. Another study on patients with ulcerative colitis agreed with our findings and in particular: clinical non-responders have significantly higher fecal concentrations of IFX after the first day of treatment than patients with clinical responses [60].
The role of TNF α agents in the pathogenesis of IBD has been well established and therapeutic interventions aimed to block or to inhibit pro-inflammatory cytokines in IBD shows good clinical results. Nevertheless, although anti-TNF agents confer remarkable therapeutic benefit compared to other available therapeutic modalities, about 30% of patients have an incomplete response, and a small percentage have no response to TNF α inhibitors. Deeping into the main mechanisms of action and loss of response to infliximab, we focused on the division between central and peripheral mechanisms, which seem to be crucial to better understand the relationship between biologic or environmental factors involved in this process. The increasing interest and the growing knowledge of the pharmacokinetics and pharmacodynamics of the infliximab could allow and give the clinician the rationale for dosage adjustments and shift to other biologic therapy.
Further studies are needed to understand if infliximab is acting primarily through local or systemic action, to find out if the loss of response is due to the characteristics of the intestinal mucosa or less, and also to perceive what role does the individual predisposition and identify biological target easily measurable. Acquire more information about pharmacokinetic and pharmacodynamic properties will showing new clinical and therapeutic perspectives about the utilization of infliximab in IBD patients.