Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Research Article - (2018) Volume 7, Issue 1
Keywords: Biochemical; Microorganisms; Antibacterials; Tetrazoles; Imidazoles
NBS: N-Bromosuccinimide; dap B: Dihydrodipicolinatereductase (EC 1.3.1.26); dap: F: Diaminopimelateepimerase (EC 5.1.1.7); dap: A: Dihydrodipicolinate synthase (EC 4.2.1.52); ddh: Diaminopimelate dehydrogenase (EC 1.4.1.16); Lys: A: Diaminopimelate Decarboxylase (EC 4. 1.1. 20).
Antibiotics are the compounds that factually ‘against life’ and interfere with structure or biochemical process that is necessary for survival and growth of microorganisms by least harm to the mammals. We live in modern epoch where antibiotic resistance has spreading at an alarming rate. Recently there is an improved interest towards development of new antibacterial agents due to emergence of newer pathogenic bacterial strains showing high resistance towards antibiotics of last resort like vancomycine [1]. Vancomycine has been used from past 30 years without resistance, evolution of vancomycin-resistant enterococci (VRE) and vancomycine resistance Staphylococcus aureus (VRSA) takes attention of scientist to develop newer agent which can combat with pathogenic bacterial strains [2]. Decline in research by medical and pharmaceutical companies from last few years which results into shortfall in developing newer agents to fight present threat of drug resistance. There is continuous need to develop antibiotics that interact with crucial mechanisms. Such compounds should be targeted toward proteins that are vital for bacterial survival but not there in mammals. L-lysine biosynthesis meets both of these criterions, presenting multiple targets for novel antibacterials. Lysine has been known to be an essential amino acid required in protein synthesis and a component of peptidoglycan layer of cell walls in gram positive bacteria as shown in Figure 1. Lysine biosynthesis also produces immediate precursor meso; DAP which is a part of peptidoglycan in gram negative bacteria and mycobacterium [3]. Substrate-based inhibitors of enzymes of DAP pathway have been reviewed and inhibitors that allow better understanding of enzymology on targets. It provides insight for designing of new inhibitors is discussed in this work. Enzymes involved in this pathway may be viable targets and shall be supportive to develop novel antimicrobial drugs. DAP is a symmetrical α,α-diaminodicarboxylic acid and exists in three stereo isomeric forms as shown in Figure 2. Among these meso-DAP and (S,S)- DAP serves as precursors in the biosynthesis of L-lysine [4].
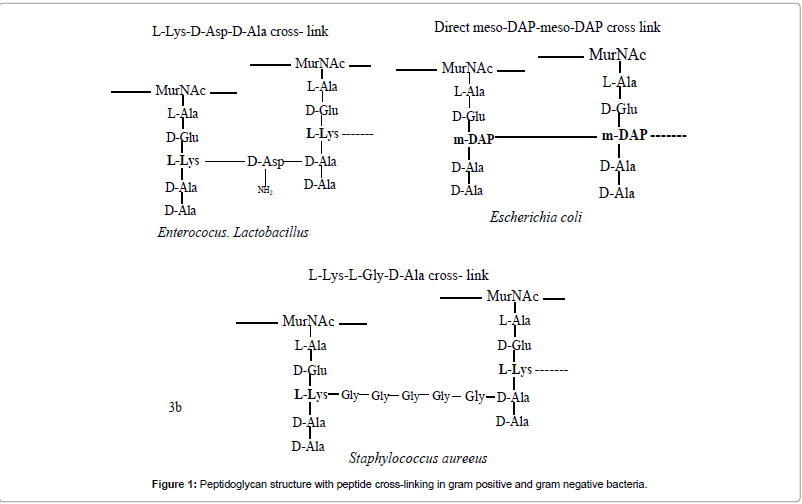
Figure 1: Peptidoglycan structure with peptide cross-linking in gram positive and gram negative bacteria.
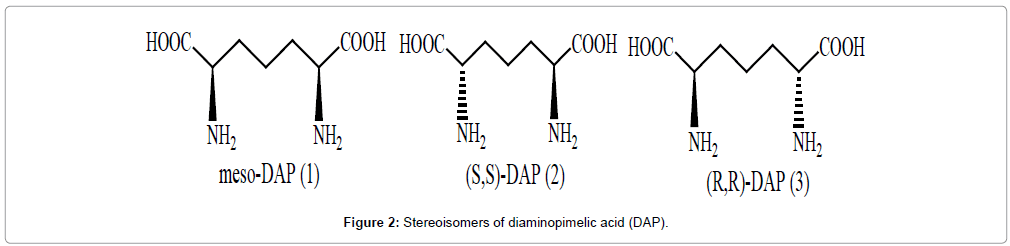
Figure 2: Stereoisomers of diaminopimelic acid (DAP).
However, only a few studies concerning the target properties of this amino acid in antibacterial drug design were published. Two new compounds (4 and 5) were planned as per SAR of reported analogues and enzyme active site study [5]. In this paper, DAP Analogues ingenuous as one terminal carboxyl group replaced with five membered heterocycle moieties containing secondary amine and central carbon replaced with sulfur were synthesized and examined as inhibitors of DAP enzymes. The amino acids were designed so that they would be reversible or irreversible inhibitors of enzymes of the lysine pathway. Compounds were subjected to in-silico studies which showed that these act as dapB, dapF, dapA and ddh inhibitors. Thus, all of these analogues were found to be the potent inhibitors. It is surmised that these heterocyclic analogues of DAP might be converted to a tight-binding transition state at the active site of these enzymes and exhibit excellent competitive inhibitory activity [6]. Toxicity risk assessment studies were conducted by using Osiris property explorer where synthesized analogs did not show any of studied toxicities.
A structure-based drug design approach has been used by analyzing previously existing analogs of DAP and active site study. The purpose of study was used to increase binding affinity against the target and correspondingly increase the cellular potency of inhibitors. Our interest in these heterocyclic analogues especially imidazole and tetrazole has risen from their potential biological activities such as antibacterial, fungicidal or herbicidal [7]. These heterocyclic analogues are able to mimic some important aspect of protein structure or their function and their three dimensional structure can behave as bioactive molecules. Such reviews motivated us to design heterocyclic analogue of DAP (4 and 5) by replacing carboxyl moiety with heterocycles of acidic characteristics (Figure 3). Tetrazoles and imidazoles have been widely used as carboxylic acid pharmacophore, especially in designing of angiotensin II, histamine antagonists; we apply this theory for designing of inhibitors. Tetrazole is metabolically stable and a close similarity between the acidic character of tetrazole and carboxylic acid has inspired medicinal chemists to synthesize substituted tetrazoles as potential medicinal agents. These heterocycle undergoes special arrangement of certain functional groups in 3 dimensional space and their electron density recognizes the site of these groups rather than the structure of entire drug molecule that result in interaction with biological system [8]. Heterocyclic ring amongst drug means that this moiety of necessity constitutes part of pharmacophore and molecule with certain structural features would elucidate a specific biological response.
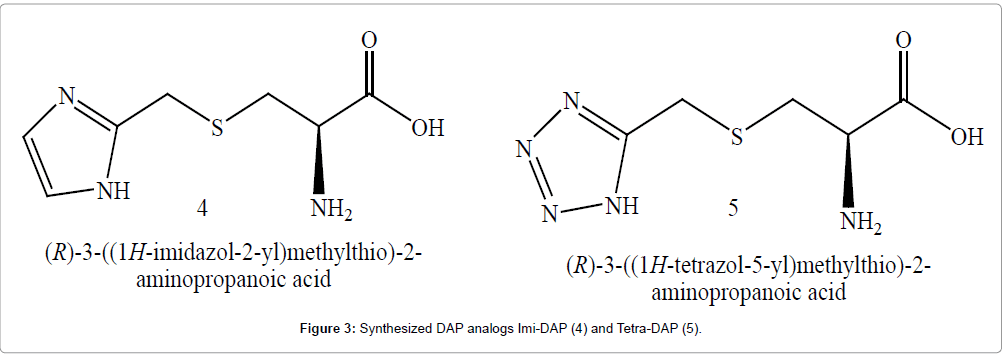
Figure 3: Synthesized DAP analogs Imi-DAP (4) and Tetra-DAP (5).
Imidazole is an important class of heterocycle that include both biological and chemical interest because constituents several natural compounds like histamine, histidine, biotin, alkaloids, nucleic acid and being a polar, ionisable aromatic compound [9]. In view of such reports current study was design to evaluate the antimicrobial activity of imidazole analogue of DAP (4). Imidazole was first synthesized by Heinrich Debus in 1858, using glyoxal and formaldehyde in ammonia and it was produces relatively low yield. Significant changes made by Radiszewski, utilizes ammonium acetate as continues source of ammonia which is generated during the heating and produces imidazole in high yield. Imidazole is 5-membered planar ring and monoacidic base having an ability to form crystalline salts with acids. Tetrazoles is rhetoric class of heterocycle with a wide range of applications viz, organo-catalysis, transition-metal catalysis, propellants, explosives, and perhaps most commonly non-classical isosteres of carboxylic acids in medicinal chemistry. The classic method of synthesizing tetrazoles from nitriles uses sodium azide in acceptable yield and purity. This also reacts on sterically hindered nitriles to produce 5-substituted-tetrazoles. However, simple route reported by Finnegan adopted for the preparation of 5-substituted-tetrazoles. Compound was cyclized using sodium azide, acetonitrile in DMF and Zinc chloride as catalyst. The main attentions are given to the problems of regioselective functionalization of the tetrazole ring and the development of convenient methods for the synthesis of C-substituted tetrazoles. Mechanism follows complex formation of acid with transition metal salts and attack of activated nitrile to form tetrazoles via formal [2+3] cycloaddition of azides and nitriles which is in transition state.
Selective protection and deprotection of functional groups is one of the major issues in multistep synthetic strategies of organic compounds. In particular, amine groups (NH) are targets for selective protection, because often selectively accessible NH-groups are required for reaction and pharmacological effect. Many NH-protecting groups are known and their ability to protect primary and secondary groups was found with variety of protection protocol. As a part of our research work, we required the selective protection of amine group (NH). A literature revealed that only few reports on the regioselective protection of amine group are available in the world of science are 9-Fluorenylmethyl carbamates (Fmoc-NR2), t-Butyl carbamates (BOC-NR2), Benzyl carbamates (Cbz- NR2), Acetamide (Ac-NR2), Phthalimide, p-Toluenesulfonamide (Ts- NR2) used to protect selectively amine. We supposed that trityl group meets our requirements sterically demanding protection selective for amines, stable under acidic condition and deprotected compound in good yield. Based on experience and literature we turned to scope of regioselective protection and subjected a heterocycles to our protection protocol. The trityl protecting group readily removed using HCI in THF. We selected the trityl group to protect the acidic N-H of 5-methyl tetrazole (15) and 2-methyl imidazole (9) for several reasons. Apart from this trityl chloride used as Organocatalyst by in situ formation of trityl carbocation with inherent instability and efficiently promotes the cross-aldol condensation reaction between cycloalkanones and arylaldehydes in solvent-free as well homogeneous media to afford α,α’- bis(arylidene) cycloalkanones in high yields. Bromination amply Wohl- Ziegler Reaction in which NBS is a valuable reagent for bromination, specific for allylic or benzylic position and addition follows free radical mechanism in which NBS serves as slow and continuous source of bromine. NBS used for brominating position α to the carbonyl or double bond, α-Bromination is first step of introducing a hetero-atom so as to provide additional conjugation and for generating stabilized carbon radicals or carbanions. Environmentally hazardous bromine has given way to NBS as a user friendly reagent and by product succinimide can be recycled.
Multicomponent reactions (MCRs) drawn great concern enjoying an outstanding status in modern organic synthesis and medicinal chemistry because one-pot processes bringing together three or more component demonstrate high atomic economy, high selectivity and emergence of powerful tools for drug discovery. As a part of our ongoing efforts to progress of one-pot synthesis for substituted heterocyclic through MRCs, we have discovered an efficient and environment friendly method for the synthesis of methyl substituted imidazole (9). Our effort to develop Brønsted acid catalyzed synthetic methodologies for synthesis of substituted imidazole by four component one-pot cyclocondensation of glyoxale (6) with Acetaldehyde (7), ammonium acetate (8) and Ethanol as solvent in presence of PTSA viz. non-toxic and inexpensive catalyst under thermal condition (Scheme 1). Microwaveassisted organic synthesis (MAOS) is new and quickly growing area in organic chemistry. In many cases reactions that normally require many hours at reflux temperature under classical conditions can be completed within few minutes or even seconds in a microwave oven at comparable reaction temperatures. Highly efficient one-pot 5-substituted tetrazole (15) synthesized by the cyclo-condensation of sodium acid (NaN3), Acetonitlile (13) in DMF and zinc chloride as catalyst under catalytic microwave system (Scheme 2). The purpose of developing MAOS for 5-substituted tetrazole (15) to provided high yields and to minimize energy requirement as compare to conventional method. To the best of our knowledge Microwave-assisted version of this reaction has not been described. Although several papers describing other microwaveassisted synthesis of N-substituted and C-substituted Tetrazole have been published, 15 and 9 were reacted under phase transfer conditions with trityl chloride to provide the product (10 and 16) in 75% recrystallized yield as a single regioisomer and deprotected using 10% HCL and 2 N NaOH in THF (Schemes 1 and 2). The resulting compound provides the side for attachment of hetero-atom like bromine to give 16 and 10 in good yield. Significantly, only a single regioisomer formed in protection step. This simplifies subsequent synthetic operations which might be carried out prior to tetrazole and imidazole deprotection. Wohl-Ziegler Reaction adapted for bromination to introduce hetero atom to α-substituted methyl through free radical process in which NBS as source of bromine [10]. Protected derivative 16 and 10 was then treated with NBS to offer a 5-bromomethyl substituted tetrazoles (17) and 2-bromomethyl substituted imidazole (11) (Schemes 1 and 2). 11 and 17 under goes final conjugation reacting with thiol group of α-amino acid cysteine (13) and susceptible to oxidation in presence of K2CO3 offered N-trityl protected analogues 18 and 12. The desired 5-alkylated tetrazoles (5) and 2-alkylated imidazole (4) were readily separated from residual trityl chloride by extraction of reaction mixture with base, taking 5 and 4 into the aqueous layer. Subsequent washing of aqueous layer with ethyl acetate removed trityl alcohol by products. Finally, reacidification and extraction with ethyl acetate provided desired products free from trityl contamination. To this end we chose the trityl-protecting amino group since this prevent partial racemization of N-protected heterocycles.
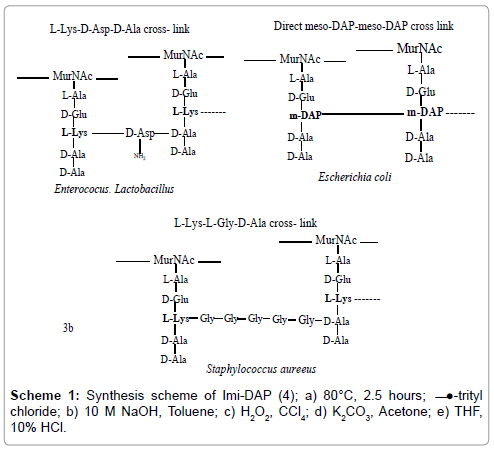
Schemes 1: Synthesis scheme of Imi-DAP (4); a) 80°C, 2.5 hours; ̶ ̶●-trityl chloride; b) 10 M NaOH, Toluene; c) H2O2, CCl4; d) K2CO3, Acetone; e) THF, 10% HCl.
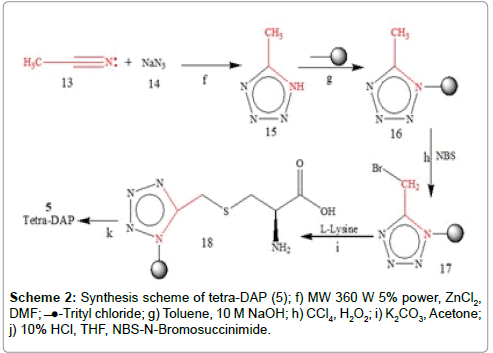
Schemes 2: Synthesis scheme of tetra-DAP (5); f) MW 360 W 5% power, ZnCl2, DMF; ̶ ̶●-Trityl chloride; g) Toluene, 10 M NaOH; h) CCl4, H2O2; i) K2CO3, Acetone; j) 10% HCl, THF, NBS-N-Bromosuccinimide.
Antimicrobial potential
Antibacterial results shown in Table 1 revealed that, the target compounds (4-5) active against both Gram-positive and Gramnegative bacteria. Compounds shows broad antimicrobial spectrum by effectively inhibiting growth of strains including B. subtilis, B.megatorium, E. coli and P. aeruginosa and in-vitro comparison with their corresponding standard drug. Individual minimal inhibitory concentration (MIC, μg/mL) represent inhibition of growth, displays bacteriostatic effect. Compounds also evaluated for bactericidal effect and concentration at which these agents kill the microorganism called minimal bactericidal concentration (MBC, μg/mL). Optical density readings of growth at 24 hours showed that these synthesized compounds (4-5) showed same degrees of MIC against test microorganisms. Turbidity was measured with the help of colorimeter at about 620 nm. Calorimetrically it was found that concentration of MIC were 80 μg/mL for DAP analogs 4 and 5 exhibit good activities against B. subtilis, B. megatorium, E-coli and P. aureogenosa. MIC and higher concentration solution used to determine MBC by observing the growth of microorganism on culture media after 24 hours at 37°C. Compound shows varying degree of MBC on above mentioned trains. Compound 5 having good MBC value against B. subtilis MBC=90 μg/ mL, B. megatorium MBC=90 μg/mL, E-coli MBC=100 μg/mL and P. aureogenosa MBC=110 μg/mL while the compound 4 was slightly higher value of MBC against B. subtilis, B. megatorium MBC=100 μg/ mL. The zone of inhibition (ZOI) of DAP analogs and ciprofloxacin was studied by cup-plate agar diffusion method at concentration of 100 μg/ mL and 200 μg/mL on culture media. The activity was observed after 48 hours at 37°C on shaking incubator. Compound 4 and 5 shows better ZOI, which conform their effectiveness against test microorganism. These ZOI were measured in mm (shown in Table 1) and respective graphs were plotted (Figure 4). Compound 4 and 5 synthesized by the isostearic replacement of carboxylic group with five member tetrazole and imidazole which contain one acidic replaceable hydrogen and these exists in two equivalent tautomeric forms, 1H-imidazole/3H-imidazole and 1H-tetrazole/3H-tetrazole, displayed significant antibacterial efficacy. Especially, these analogs give comparable as well as satisfactory activities in comparison with clinical drug ciprofloxacin at the concentrations of 70-80 μg/mL against all tested bacteria. Particularly compound 5 with tetrazole moiety show slightly higher potency than 4. Compound 5 has strongest inhibition against B. subtilis which was approximately equal potent than ciprofloxacin at both concentration of ZOI. This result suggested that the existence of tetrazole and imidazole as carboxylic acid pharmacophores would have special importance in medicine due to their bulky nature they imparts high lipophilicity which helpful for biological transportation and distribution. In order to briefly examine previously reported analogs in which replacement of both carboxyl’s of DAP with phosphonic acid, bis-phosphonate analogue P-DAP isomers was also synthesized as a stereoisomeric mixture (DD, LL and meso). Free phosphates are known to especially resistant to un-assisted crossing of cell membranes. It was also observed that microorganisms were sensitive to 4 and 5. Furthermore, we thought that these analogs might be active against resistance bacterial strain like VRSA, VRE and MRSA because all type of bacteria utilizes important biochemical process DAP pathway that is synthesis of lysine. These resistance bacterial strains was known to be most distraught and virulent organism that caused a broad array of problems to hospitalized and community acquired patients, and showed multi-drug resistance to numerous currently available agents. All these results will be a good starting point to optimize structure to obtain potent antimicrobial agents with these simple scaffolds. Further researches, including in vivo bioactive evaluation, incorporation of different linkers (alkyl, aryl and heterocyclic moieties) and diverse heterocyclic azoles (pyrazole, oxazole, carbazole, benzimidazole, benzotriazole etc.) into DAP backbone as well as various functional groups (ester, ketone, amide ones and metal, etc.). Nevertheless, substitution of heterocyclic ring in DAP causes remarkable enzyme inhibition properties for DAP pathway and potentiate antimicrobial properties of newly synthesized analogs. The structural necessities for substrate recognition are quite strict which would bind with correct α-amino acid functionality at the active site and would place the stereo chemically correct polar carboxylic group at the distal recognition site through which it acts as a substrate with effective binding. The distal (non-reacting) α-amino acid site must have L-configuration and both carboxyl and both amino groups are necessary for substrate recognition and transformation (α-hydrogen exchange) by dapF from E. coli. The substitution of carboxyl in DAP by a heterocyclic ring may alter the pKa of the adjacent α-hydrogen considerably. It is interesting that the distal (non-reacting) recognition sites in dapF and lysA prefer to bind the natural α-amino acid moiety and demand greater structural fidelity than protein groups which binds reacting terminus of the DAP molecule [11]. Moreover, the antimicrobial potency for 4 and 5 depend on α-amino acid like arrangement at one terminal of substrate restricts us to utilize L-cysteine as a precursor for synthesis and also depend upon 3 to 4 carbon lengths of aliphatic chains from α-amino acid distal recognition site. The activities might be decrease with increase or decrease of aliphatic chain length. Central carbon of DAP exchanged with sulfur which cause change in bond length and bond angle of synthesized analogs may leads to affect enzyme binding properties. Obviously, the bridged linker central sulfur have large effect on their antimicrobial efficiency, further work is essential in order to deduce the structure–activity relationship. This transformation of carboxylic group into acidic heterocycle might modulate the lipid/water partition coefficient which affect their diffusion in bacterial cells as well as interaction with bacterial cells or tissues and thereby improve the pharmacological properties. Synthesized compound were confirmed by IR, MS, melting point, elemental analyses and all the reactions optimized by TLC. The in vitro antibacterial evaluation showed that synthesized compounds (4-5) effectively inhibit the growth of all tested bacteria.
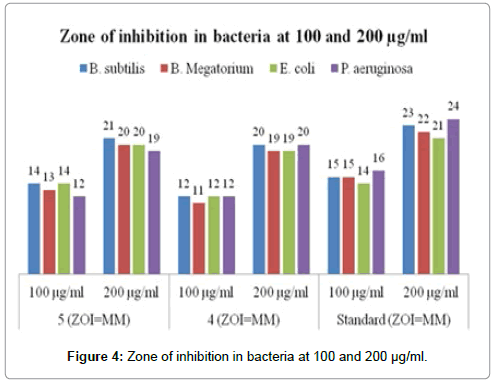
Figure 4: Zone of inhibition in bacteria at 100 and 200 μg/ml.
| Microorganism | 5 | 4 | Standard | 5 (ZOI=MM) | 4 (ZOI=MM) | Standard (ZOI=MM) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | 100 µg/ml | 200 µg/ml | 100 µg/ml | 200 µg/ml | 100 µg/ml | 200 µg/ml | |
| B. subtilis | 80 | 90 | 80 | 100 | 70 | 80 | 14 | 21 | 12 | 20 | 15 | 23 |
| B. megatorium | 80 | 90 | 80 | 100 | 70 | 90 | 13 | 20 | 11 | 19 | 15 | 22 |
| E. coli | 80 | 100 | 80 | 100 | 80 | 90 | 14 | 20 | 12 | 19 | 14 | 21 |
| P. aeruginosa | 80 | 110 | 80 | 110 | 70 | 80 | 12 | 19 | 12 | 20 | 16 | 24 |
Table 1: Minimum inhibitory concentration (MIC) and zone of inhibition (ZOI).
Present work draw attention to novel chemical analogs of DAP (4 and 5) acting as antimicrobial through enzyme inhibition especially involved in lysine biosynthesis known as DAP pathway. Development of antibacterial agents by this target realizes that there is continuous need to develop innovative analogs to combat with the resistance imposed by bacteria. Such newer analogs shall be able to target and inhibit enzymes or protein biosynthesis which is crucial for the survival. Selective inhibition of enzymes of DAP pathway by appropriate substrate analog to develop new drugs that are more effective to resistance bacterial strains. An enzymes involved in this pathway may be a feasible targets for drug discovery towards protein and enzyme inhibition in this way generate newer avenues in treatment of diseases using antimicrobial therapy. Such drugs shall be able to stand in the market with good therapeutic results for a longer duration of time and also with a higher success rate without fall down by bacterial resistance. A study at a deeper level is needed to confirm the quantitative extent of inhibition.
The authors are thankful to the Principal, Government College of Pharmacy, Aurangabad, Maharashtra, India for providing research facility to carry out this research work.