Chemotherapy: Open Access
Open Access
ISSN: 2167-7700
ISSN: 2167-7700
Research Article - (2023)Volume 11, Issue 4
Objective: RNA N6-methyl Adenosine (m6A) plays a key role in tumor development. However, its role in breast cancer has not been fully elucidated.
Methods: In this study, 23 m6A RNA regulatory factors were systematically analyzed from the TCGA database to analyze the relationship between m6A and breast cancer by bioinformatics. Then, we further detected the expression of m6A by immunohistochemical method, and analyzed the relationship between it and the prognosis of breast cancer.
Results: We found increased expression of 6 regulators in breast cancer patients. We further studied the effect of abnormal expression of CBLL1, YTHDC1, METTL14, HNRNPA2B1, RBMX, and HNRNPC on the survival time of breast cancer. In addition, we also found that the abnormality of the M6A regulatory factor is closely related to immune infiltration and genetic changes.
Conclusion: Our findings suggest that the expression of m6A regulators in breast cancer has important clinical diagnostic significance and that abnormal expression may be associated with poor clinical prognosis in BC. This helps to provide us with a new direction of targeted therapy for breast cancer.
m6A; RNA methylation; Breast cancer; TCGA; Immune infiltration
m6A: N6-methyl Adenosine; TCGA: Tumor Cancer Genome Atlas; GEO: Gene-Expression Omnibus; ICGC: International Cancer Genome Consortium; aDC: activated DC; iDC: immature DC; pDC: plasmacytoid DC; Tcm: T central memory; Tem: T effector memory; Tfh: T follicular helper; Tgd: T gamma delta; DAVID: Database for Annotation, Visualization and Integrated Discovery; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; ROC: Receiver Operating Characteristic; BCSC: Breast Cancer Stem Cells; VIRMA: Virilizer Homolog; TIL: Tumor-Infiltrating Lymphocytes; TAM: Tumor-Associated Macrophages; MDSC: Myeloid Suppressor Cells
According to the latest report of the World Health Organization, the incidence of breast cancer is increasing year by year. And the incidence of breast cancer has surpassed that of lung cancer and has become the highest incidence of malignant tumors, accounting for 24.5% of female malignant tumors. It has become the highest and most common carcinoma among women [1,2]. BRC is also the leading cause of cancer-related death for women in the vast majority of countries [3]. Despite significant advances in treatment in recent years, the prognosis for breast cancer remains poor. Therefore, further research is needed to determine the underlying mechanism of breast cancer occurrence and development.
Epigenetic irregularity is a consistent feature of many cancers [4]. In addition to well-known classical epigenetic regulatory mechanisms such as chromatin remodeling, DNA methylation, and histone modification, RNA N6-methyl Adenosine (m6A) is also considered as a new epigenetic regulator, and has been considered one of the hottest research projects in the epigenetic area [5]. RNA N6- methyl Adenosine (m6A) [6], which happens at the N6 position of adenosine, is an internal modification prevalent in mammalian mRNA and non-coding RNAs, and it plays the ubiquitous role of "writer", "eraser" and "reader" in RNA processing [7], and it influences mRNA splicing, export, localization, translation, decay, and stability. The writer complex contains the methyltransferases METTL3 and METTL14, the METTL3 adaptor WTAP, and other related proteins, including KIAA1429, RBM15/15B, and ZC3H13 [8]. In the METTL3-METTL14 methyltransferase domain complex, METTL3 acts as the catalytic subunit, whereas METTL14 provides an RNA-binding scaffold that activates and enhances the catalytic activity of METTL3 [9]. WTAP is catalytically inactive but can interact with METTL3 and METTL14 to regulate the m6A level of RNA transcription [10]. Two different enzymes are known to be involved in the m6A elimination process: FTO and ALKBH5 [11]. However, it was found that FTO preferentially demethylates m6A, an m6A-related nucleoside that is present in mRNA in high abundance [12]. ALKBH5 has been found to be a major m6A demethylase [13]. A growing body of evidence suggested that m6A regulates gene expression and plays a vital part in cancer through a variety of mechanisms. New evidence showed that m6A modification is associated with tumor proliferation, glycolysis, apoptosis, and metastasis [14]. Abnormal expression of M6A methylation regulatory factors can also lead to cancer. However, M6A modification can play both carcinogenic and anticancer roles in malignant tumors. Such as ALKBH5 and METTL3 play a distinct role in different tumor types [15-17]. m6A regulators performed variant functions to different tumor types showing that the regulation of m6A methylation modification levels is absolutely complex. Found by reading relevant literature,m6A plays an significant role in the tumor immune microenvironment, which in turn affects the occurrence, development and treatment sensitivity of tumors. A study has shown that key functions in regulatory T cells have a clear m6A mRNA dependence, making them potentially important targets in anti-tumor immunotherapy. This is shown in Table 1; More and more studies have shown that m6A is associated with the development of breast cancer. Closely related to the exhibition. m6A regulatory protein is heavy in the development of cancer. To adjust factor, its level of expression is often directly determines the swollen tumor pathological process. Although the evidence revealed m6A RNA methylation involved in the onset and development of cancers, the specific role of m6A in breast cancer has not been fully demonstrated [35].
| m6A regulators | m6A Modification type | Expression trend | Target gene | Function | Reference |
|---|---|---|---|---|---|
| METTL3 | Writer | ↑ | HBXIP | Promote cell proliferation | [18] |
| ↓ | Bcl-2 | Inhibition of apoptosis | |||
| METTL14 | Writer | ↑ | has-miR-146a-5p | Promote cell proliferation, migration and invasion, promote cell colony formation, promote cell cycle | [19,20] |
| ↓ | CXCR4, CYP1B1 | Inhibit clonogenesis and cell viability | |||
| WTAP | Writer | ↓ | - | - | [20] |
| VIRMA/KIAA1429 | Writer | ↑ | CDK1 | Promote cell proliferation and metastasis | [21] |
| RBM15 | Writer | ↑ | - | - | [22] |
| RBM15B | Writer | ↑ | - | - | |
| CBLL1 | Writer | - | ERa | Inhibit cell proliferation and migration | [23] |
| FTO | Eraser | ↑ | BNIP | Promote cell proliferation and metastasis, promote cell clonal formation ability, increase ATP production, promote glycolysis and lactic acid formation | [24,25] |
| P13K/AKT | |||||
| ALKBH5 | Eraser | - | NANOG | Promote cell viability, clone formation and metastasis | [26] |
| IGF2BP1/IMP1 | Reader | ↑ | UCAI | Promote cell proliferation, reduce invasion and metastasis | [27] |
| ↓ | c-MYC, IGF-II | Inhibition of tumor growth | |||
| IGF2BP2/IMP2 | Reader | ↑ | PR | Promote migration, migration and growth, promote self-renewal | [28] |
| RPSAP52 | |||||
| IGF2BP3/IMP3 | Reader | ↑ | WNT5B | [29] | |
| BCRP | Promote cell proliferation, invasion and metastasis, promote cell drug | ||||
| CD44 | Resistance, promote the stem-like properties | ||||
| CERS6 | |||||
| EIF3H | Reader | ↑ | YAP | Promote cell migration and metastasis | [30] |
| HNRNPC | Reader | ↑ | - | Promote cell proliferation | [31] |
| HNRNPA2B1 | Reader | ↑ | STAT3, EKR1/2 | Promote cell proliferation and reduce cell apoptosis | [32,33] |
| ↓ | ERK-MAPK/Twist | Inhibit cell metastasis | |||
| GR-beta/TCF4 | |||||
| PFN2 | |||||
| YTHDF1 | Reader | ↑ | eIF3C | The translation efficiency of mRNA modified with m6A methylation was improved | [34] |
Note: (↑) High; (↓) Low.
Table 1: List of the expression and function of m6A regulatory proteins in breast cancer.
Therefore, we systematically analyzed 23 m6A methylation regulators and examined their expression of them in breast cancers and normal controls based on open biological data. This study used the data in the Tumor Cancer Genome Atlas (TCGA) database to analyze the expression of m6A methylation regulator in breast cancer and its relationship with clinicopathological characteristics and we also used bioinformatics methods to predict the potential functions of these m6A regulators. Finally, we analyzed the critical m6A methylation regulators and uncovered the m6A-related biological mechanism in breast cancer.
Data source
Public gene expression data, copy number variation data, RNA methylation data, and clinical annotations were collected from the Tumor Cancer Genome Atlas (TCGA) database, the Gene- Expression Omnibus (GEO), and the International Cancer Genome Consortium (ICGC). including 1099 cancer samples and 114 normal samples. Exclusion criteria: (1) there is no complete RNA sequencing data in the sample; (2) the sample is not before treatment; (3) advanced breast cancer. Finally, 1092 cases of breast cancer and 113 cases of normal breast tissue were included.
Selection and differential expression analysis of m6A methylation regulators
According to the related literature [36,37], we selected 23 m6A methylation regulators that regulate RNA methylation, including 10 writers, and 2 erasers, 11 readers. The differential expression analysis of 23 m6A regulators in breast cancers and normal controls was compared by the "limma" package in R, a heatmap was assessed by the"Complex Heatmap" package.
Expression analysis
We used UALCAN (http://ualcan.path.uab.edu/index.html) to confirm the correlation between m6A regulators and clinical parameters in breast cancer [38].
Gene alteration analysis of m6A regulator in breast cancer
CBioPortal (http://www.cbioportal.org/) is an open-access website to explore, visualize and analyze multidimensional cancer genomics data. We used this website to analyze the genetic alterations of m6A regulators in breast cancer.
Immune response analysis
The correspondence of m6A regulators with levels of immune cell infiltration and Immune infiltration score were detected. Immune infiltrating cells16 include: activated DC (aDC); B cells; CD8 T cells; Cytotoxic cells; DC; Eosinophils; immature DC (iDC); Macrophages; Mast cells; Neutrophils; NK CD56bright cells; NK CD56dim cells; NK cells; plasmacytoid DC (pDC); T cells; T helper cells; T central memory (Tcm); T effector memory (Tem); T follicular helper (Tfh); T gamma delta (Tgd); Th1 cells; Th17 cells; Th2 cells; Treg. immune infiltration score [39] include: Stromal Score, ESTIMATE Score, Immune Score. Then, we assessed the correlations between the expression of m6A regulators and Immuno regulators (including Immuno inhibitors, Immuno stimulator, and MHC molecules) by using TISIDB database (http://cis.hku.hk/TISIDB/).
Survival analysis
GCSALite mRNA expression module calculates the genomic differential expression between different cancers according to TCGA expression data. The results of module analysis provide differential expression, survival analysis, and subtype analysis [40]. For expression survival analysis, we use 33 cancer types of clinical tumor data, some uncensored data was left out. Merging mRNA expression and clinical survival data by sample barcode, we use median RSEM value to divide tumor samples into high and low groups. Then, we use the R package survival to fit survival time and survival status with two groups. We calculate the Cox Proportional- Hazards model for every gene and draw Kaplan-Meier curves with log-rank tests for every single gene. The genes with Kaplan-Meier log-rank test p-value less than 0.05 would be retained and gathered into the final figure.
The correlation between survival time and expression of m6A regulators in breast cancer was measured by using the K-M map of the website (http://kmplot.com/analysis/) and determined by the endpoint reported before.
GO and KEGG analysis
Using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://davidd.ncifcrf.gov/), we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis based on CO expressed genes. Select the critical value p<0.05 as the critical value.
Statistical analysis
All statistical analyses were performed using R software (version 4.0.5). The Wilcoxon's test was applied to contrast the expression of m6A regulators between cancer and normal tissues. Lasso regression was performed by the " glmnet" package in R. The chisquare test was used to compare the relationship between m6A and Immune Response. The validation of the diagnostic models was assessed by the Receiver Operating Characteristic (ROC) curve. For all the analyses, a P-value less than 0.05 was regarded as statistically significant.
Abnormalities of different types of m6A regulators are associated with human cancers survival
To assess the functional role of the m6A regulators in human cancers, we used the GCSALite mRNA expression module based on the TCGA expression data to analyze the association between Abnormalities of different types of M6A regulators and survival time and survival status in human cancers. As shown in Figure 1, the dot represents the gene that affects the survival of the cancer types, the p-value is the Kaplan Meier P-value. The dot color indicates the worse of the high or low expression in the cancer types. The larger the dot, the greater the genetic influence. The results show that the types of genes that influence cancer survival and their expression vary from tumor to tumor. For example, there were eight genes affecting the survival of breast cancer patients, among which 4 genes such as IGF2BP1, KIAA1429, FTO, YTHDF3, METTL16, YTHDF1 were highly expressed, and 2 genes such as HNRNPC and RBM15B were low expressed. And we can find that RBM15B had a significant effect.

Figure 1: Abnormalities of different types of m6A regulators are
associated with human cancers survival. The correlation between
abnormalities of different types of m6A regulators and survival in
patients with kidney renal clear cell cancer, brain lower grade glioma,
adrenocortical cancer, breast cancer, and 25 other tumors. Note: Effect
of high exp. on survival risk 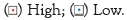
The aberrations of m6A regulators were correlated to prognosis in breast cancer
The study focused on exploring the role of m6A in the prognosis of breast cancer patients. According to whether m6A methylation was performed, the patients were divided into altered group and Unaltered group. The Altered group (n=1136) and Unaltered group (n=1933) Figure 2A, the Altered group (n=742), and the Unaltered group (n=1336) Figure 2B, were included in the TCGABRCA database. After comparison, we could conclude that the 5Aaltered group was remarkably correlated to a worse prognosis. In other words, the altered group had a shorter OS time and was more prone to relapse. According to the results of Figure 1, we know that the expression of m6A regulators is different in breast cancer, but as shown in Figures 2A and 2B, the occurrence of m6A methylation is associated with poor prognosis of breast cancer.

Figure 2: The Aberrations of m6A regulators were correlated to
prognosis in breast cancer. 
Amplification, deletion, and mutation of m6A regulators in breast cancer
In this study, we were queried 6110 patients/6437 samples in 8 studies to explore genetic changes. As present in Figure 3, we found varying degrees of genetic change in 23 m6A regulators, including, METTL3, METTL14, WTAP, ZC3H13, RBM15, RBM15B, VIRMA, METTL16, FTO, ALKBH5, YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1, IGF2BP1, IGF2BP2, IGF2BP3, RBMX, HNRNPC, ZCCHC4, CBLL1. We revealed most of the m6A regulators were amplified, deleted, mutated in BRCA, among which VIRMA displayed the highest incidence rate (17%). We can also find that VIRMA is prone to Amplification, followed by YTHDF3. FTO is prone to Deep deletion.

Figure 3: Amplification, deletion, and mutation of m6A regulators in
breast cancer. Querying 6110 patients/6437 samples in 8 studies. 
Expression profile of m6A regulators in breast cancer
The level of m6A regulators in TCGA was presented in Figure 4A. We compared the expression of 23 m6A regulators in cancer and normal tissues. There was significant differential expression of 14 genes showed in the heat map Figure 4A, vio-plot revealed the same result Figure 4B. The m6A regulators include Writer: METTL14, METTL16, WTAP, VIRMA, KIAA1429, ZC3H13, RBM15; Erasers: FTO; Readers: YTHDC1, YTHDF1, HNRNPA2B1, HNRNPC, IGF2BP1, IGF2BP2, IGF2BP3, ZCCHC4. Among them, VIRMA/ KIAA1429, YTHDF1, HNRNPA2B1, HNRNPC, IGF2BP1, and IGF2BP3 were significantly up-regulated in breast cancer tissues. However, the expression of METTLE14, METTL16, WTAP, FTO, ZC3H13, YTHDC1, IGF2BP3, ZCCHC4 was reduced to varying degrees compared with normal samples. Meanwhile, there was no difference in METTL3, ALKBH5, RBM15, RBM15B, YTHDF2, YTHDF3, YTHDC2, RBMX, CBLL1 expression between normal and tumor samples.

Figure 4: Expression profile of m6A regulators in breast cancer (A,B).
The expression levels of 6 m6A regulators showed increased expression
in EC samples, and 8 regulators showed reduced expression. 
Dysregulation of m6A is correlated with shorter OS times in breast cancer patients
In this study, we used the k-M curve to detect the role of each m6A regulator in the prognosis of breast cancer patients (Figures 5A-5F). We found that high CBLL1 Figure 5A, HNRNPA2B1 Figure 5B, HNRNPC Figure 5C, METTLE14 Figure 5D, RBMX Figure 5E, YTHDC1 Figure 5F expression has a significant impact on the poor prognosis of breast cancer patients. This suggests that m6A plays an essential role in breast cancer outcomes. We should note that Figure 5A shows the effect of high and low gene expression on OS in breast cancers where the expression of this gene was determined.

Figure 5: Dysregulation of m6A is correlated with shorter OS times in breast cancer patients (A-F) higher levels of CBLL1 (A), HNRNPA2B1
(B), HNRNPC (C), METTL14 (D), RBMX (E), and YTHDC1 (F) were significantly associated with worse outcomes in breast cancer. 
The aberrations of m6A regulators are associated with breast cancer clinical parameters
We analyzed 6 genes, among which, compared with normal genes, HNRNPA2B1 Figure 6A and HNRNPC Figure 6B were up-regulated, YTHDC1 Figure 6C and METTLE14 Figure 6D were down-regulated, and CBLL1 Figure 6E and RMBX Figure 6F showed no significant difference.

Figure 6: The aberrations of m6A regulators are associated with breast cancer clinical parameters (A-F). (A) HNRNPA2B1; (B) HNPNPC; (C) YTHDC1; (D) METTLE14; (E) CBLL1; (F) RBMX.
The differences in gene expression among Caucasian, Asian, and African American races, clinical stage, lymph node stage, and tumor subtypes were further analyzed. HNRNPA2B1 was upregulated compared with normal expression in the race, stage 1-4, N0-N3, and tumor subtypes. It is noteworthy that the expression of N3 is relatively reduced compared with that of N0-N2. HNRNPC gene expression was higher than normal in all four comparisons. However, the expression of this gene is more significantly increased in African-American breast cancer patients and luminal breast cancer, and the expression of this gene is relatively decreased in stage 4 compared with stage 1-3, and it is also decreased in N3 compared with N0-N2.
By comparing the differences in the expression of YTHDC1 and METTLE14 in the three races, clinical stages, lymph node stages, and tumor subtypes, in each comparison, we found that all expressions were lower than in the normal sample, African Americans were significantly lower than in the other two groups of breast cancer patients, and the decline in stage 4 was greater than in the first three stages. The decrease of N3 expression was more obvious than that of N0-N2, and the decrease of triple-negative breast cancer was more obvious than that of the other two subtypes.
Among patients with abnormal CBLL1 expression, only African- American breast cancer patients showed a statistically significant decrease in CBLL1 expression compared with normal and the other two groups. Comparison with normal samples and groups showed that the expression of the RBMX gene was decreased in African-American breast cancer patients, decreased in stages 1 and 4, decreased in N3, and decreased in HER2-positive breast cancer, with statistical significance. In combination with Figures 5A and 6A, there is no significant difference in the expression of CBLL1 and RBMX in breast cancer patients compared with normal patients in general. However, when these two genes are specifically expressed, higher gene expression is associated with poorer OS.
The expression of m6a regulators correlated with the level of immune infiltration in breast cancer
By reading relevant literature, we found that M6A is associated with immunity in bladder cancer, esophageal cancer and liver cancer. Therefore, we carried out GO and KEGG analysis with the hypothesis that M6A is associated with immunity in breast cancer, and found no KEGG result. The GO results are shown in the figure. Based on TIMER database, we examined the correlation between m6A regulators and the level of immune cell infiltration in breast cancer. The results are shown in Table 2. As shown in Figure 7, M6A regulators are associated with immune infiltration in breast cancer.
| Gene expression | Purity | B cell | CD8+ T cell | CD4+ T cell | Macrophage | Neutrophill | Dendritic Cell | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Paiiial cor | p | Paiiial cor | p | Paiiial cor | p | Paitial cor | p | Paitial cor | p | Paitial cor | p | Paitial cor | p | |
| CBILI | 0.071 | 0.0251* | 0.058 | 0.0689 | 0.284 | l.27E-19*** | 0.108 | 0.000824* ** | 0.196 | 5.18E-10*** | 0.16 | 7.04E-07 | 0.064 | 0.0409* |
| HNRNPA2Bl | 0.203 | 1.lIE-10*** | 0.169 | 1.0IE-07*** | 0.14 | l.22E-05*** | 0.152 | 2.22E-06 *** | 0.038 | 0.236 | 0.173 | 8.32E-08 *** | 0.135 | 2.87E-05 *** |
| HNRNPC | 0.23 | 2.08E- 13*** | 0.024 | 0.449 | 0.076 | 0.0 172* | -0.02 | 0.527 | 0.038 | 0.228 | 0.006 | 0.847 | -0.046 | 0.154 |
| l\.ffiTTLE 14 | 0.032 | 0.312 | 0.043 | 0.18 | 0.324 | 2.87E-25*** | 0.101 | 0.00164** | 0.292 | 8.14E-2 1*** | 0.201 | 3.66e-10*** | 0.095 | 3.30E-03** |
| RBMX | 0.146 | 3.48E-06*** | 0.009 | 0.788 | 0.244 | l.09E-14*** | 0.122 | 0.000104** | 0.063 | 0.0469* | 0.137 | 2.27E-05*** | 0.038 | 0.238 |
| YTHDCl | 0.087 | 0.00624** | 0.031* | 0.326 | 0.294 | 5.57E-21*** | 0.16 | 5.8 1E-07*** | 0.23 | 2.83E-13*** | 0.207 | l.28E-10*** | 0.092 | 0.00474** |
Note: *p< 0.05; **p< 0.01; ***p< 0.0001.
Table 2: The correlation between m6A regulators and the level of immune cell infiltration in breast cancer.

Figure 7: The expression of m6A regulators correlated with the level of immune infiltration in breast cancer. Based on the TIMER database, we examined the correlation between the expression of the CBLL1 (A), HNRNPA2B1 (B), HNRNPC (C), METTLE14 (D), RBMX (E), YTHDC1 (F), and the level of immune cell infiltration in breast cancer.
The relevance between m6a regulators expression and immuno regulators expression in breast cancer
To further explore the effects of m6A regulators on tumor immune response, we calculated the correlation between the expression of m6A regulators and immune regulators. As shown in Figure 8A, the expression level of CBLL1, HNRNPA2B1, HNRNPC, METTLE14, RBMX, YTHDC1 were negatively correlated with Immunoinhibitors Figure 8A, Immunostimulators Figure 8B, and MHC molecules Figure 8C expressions in breast cancer.

Figure 8: Correlations between m6A regulators expression and the expression of immuno regulators in breast cancer. The correlations between the expression of CBLL1, HNRNPA2B1, HNRNPC, METTLE14, RBMX, YTHDC1, and Immunoinhibitors (A), Immunostimulator (B), and MHC molecules (C) were calculated based on the TISIDB database.
GO and KEGG analysis
We implemented GO and KEGG enrichment analysis of CBLL1 Figure 9A, HNRNPA2B1 Figure 9B, HNRNPC Figure 9C, METTL14 Figure 9D, RBMX Figure 9E, and YTHDC1 Figure 9F through Xian tao academic website (https://www.xiantao.love/products/), to explore the functional and enrichment analysis of these m6A regulators. The results showed that CBLL1 was remarkably related to nuclear speck, methyltransferase complex, methylation, macromolecule methylation, RNA modification, RNA methylation, mRNA modification, mRNA methylation, entry of bacterium into the host cell, and so on (Figure 9A). HNRNPA2B1 was significantly related to mRNA 3'-UTR binding, spliceosomal complex, RNA splicing, mRNA splicing, via spliceosome, RNA splicing, via transesterification reactions with bulged adenosine as a nucleophile, regulation of mRNA metabolic process, regulation of mRNA processing, regulation of RNA splicing, mRNA transport and so on (Figure 9B). HNRNPC was relevant to Spliceosome, mRNA 3'-UTR binding, RNA splicing, RNA splicing, via transesterification reactions, mRNA splicing, via spliceosome RNA splicing, via transesterification reactions with bulged adenosine as a nucleophile, regulation of mRNA metabolic process, and so on (Figure 9C). METTL14 was significantly correlated with RNA splicing, RNA splicing, via transesterification reactions, mRNA splicing, via spliceosome, regulation of mRNA metabolic process, methyltransferase complex, RNA modification, RNA methylation, positive regulation of mRNA metabolic process, mRNA modification, and so on (Figure 9D). RBMX was associated with RNA splicing, via transesterification reactions with bulged adenosine as a nucleophile, regulation of mRNA metabolic process, Spliceosome, Single-stranded RNA binding, mRNA 3'- UTR binding, G-rich strand telomeric DNA binding, spliceosomal complex, catalytic step 2 spliceosome, methyltransferase complex, and mRNA splicing, via spliceosome (Figure 9E). YTHDC1 was significantly related to RNA splicing, RNA splicing, via transesterification reactions, regulation of mRNA metabolic process, regulation of mRNA processing, regulation of RNA splicing, regulation of mRNA splicing, via spliceosome, mRNA export from the nucleus, and so on (Figure 9F).

Figure 9: (A-F) GO and KEGG enrichment.
RNA molecules are an essential building block of all living organisms. These molecules act as carriers to transfer genetic information from DNA to proteins and as regulators of various biological processes [41]. RNA transcripts may undergo diverse and complex chemical modifications to tailor their structure to specific molecular functions. Indeed, more than a hundred post-transcriptional modifications have been found in cellular RNAs [42]. N6-methyl Adenosine (m6A) is considered to be the most abundant and most conserved internal modification in mRNA and long non-coding RNAs and is methylation of the nitrogen-6 position adenosine base [43]. N6-methyl Adenosine (m6A) modification was discovered and partially characterized in a great variety of cellular mRNAs in the 70s decade [44,45]. m6A represents the most abundant internal modification of eukaryotic mRNA and accounts for more than 80% of all RNA base methylations in various species. m6A mRNA methylation affects almost every stage of mRNA metabolism [46]. Research has shown that M6A methylation also regulates the expression of important genes and pathways that regulate cell fate, and dysregulation of these pathways by abnormal m6A methylation may lead to cancer [6], for example, m6A regulates the PI3K/AKT pathway during T-cell development [47-49] as well as in endometrial cancer, renal cell carcinoma, and AML [50]. In recent years, the role of the m6A regulator in breast cancer has been revealed in several previous studies. Such as aberrations of the m6A readers YTHDF1 and YTHDF3 are associated with metastasis in breast cancer patients and predict poor prognosis [51]. Altered genomic targets are associated with N6-methyladenosine in breast cancer tissue and associated with poor survival [52]. Similar results have been observed in breast cancer cells, where ALKBH5- mediated m6A demethylation of NANOG mRNA enhances Breast Cancer Stem Cells (BCSC) under hypoxia [53]. However, previous studies on the association between m6A methylation and breast cancer were mostly limited to a single molecule. Our study is the first to demonstrate that m6A regulators can be used as potential biomarkers for breast cancer prognosis and immunotherapy.
In this study, we systematically analyzed the expression of 23 m6A regulatory factors in breast cancer, and we found that abnormalities in the m6A regulatory factor in the genome are associated with breast cancer prognosis. In this study, we revealed that most m6A regulators are amplified, deleted, and mutated in breast cancer, with VIRMA having the highest incidence (11%). Virilizer Homolog (VIRMA) is also a key component of m6A methyltransferase and plays a role in the recruitment of core catalytic components METTL3, METTL14, and WTAP [54]. In TGCT, VIRMA and YTHDF3 may be prognostic factors [55]. At the same time, we found increased expression of six m6A regulators in breast cancer samples. Including VIRMA/KIAA1429, YTHDF1, HNRNPA2B1, HNRNPC, IGF2BP1, and IGF2BP3. However, the expression of METTLE14, METTL16, WTAP, FTO, ZC3H13, YTHDC1, IGF2BP3, ZCCHC4 was reduced to varying degrees compared with normal samples. Meanwhile, there was no difference in METTL3, ALKBH5, RBM15, RBM15B, YTHDF2, YTHDF3, YTHDC2, RBMX, CBLL1 expression between normal and tumor samples (Figure 4A). We looked further and found that the high expression of CBLL1, HNRNPA2B1, HNRNPC, METTLE14, RBMX, and YTHDC1 had a significant impact on the poor prognosis of breast cancer patients Figure 5A, indicating these m6A regulators play important roles in breast cancer and hold the key to the prognosis of patients. Previous studies have shown that in eukaryotes, proteins containing the YTH domain include five functional genes: YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2, which are distributed in the nucleus and cytoplasm. Proteins containing the YTH domain first recognize the m6A modification of the target RNA and then guide different complexes to regulate RNA signaling pathways, including RNA folding, RNA splicing, protein translation, and RNA metabolism [56]. Among proteins containing the YTH domain, YTHDC1 is the only one that is enriched in the nucleus. It regulates the splicing of mRNA by bridging the interaction between trans and cis-regulatory elements and combining with targeted mRNA [57]. Studies have suggested that YTHDC1 may be differentially expressed in different tumors. For example, YTHDC1 is highly expressed in colorectal adenocarcinoma, but not in rectal adenocarcinoma [58]. In gynecologic tumor cell lines, RNA splicing is altered by decreased YTHDC1 protein levels due to hypoxia [59]. The evidence indicates that YTHDC1 is associated with prostate cancer [60]. METTL14 plays a vital role in the recognition of m6A methylated RNA substrates. Studies have shown that abnormal expression of METTL14 can alter the fate of m6A-regulated transcripts. METTL14 has been reported to alter m6A levels in HCC3 [61]. HnRNPA2B1, as an RNA-binding protein, accelerates the processing of pre-mRNA by forming a complex with lncRNA during the development of cancer. HNRNPA2B1 is involved in the development of pancreatic cancer [62], prostate cancer [63], hepatocellular carcinoma [64], and lung cancer [65]. HCC patients with high cytoplasmic expression of hnRNPA2B1 showed a significantly higher incidence of low differentiation stage and lower survival rate than those with nuclear expression of hnRNPA2B1 [66-68]. In the study, increased RBMX expression was found to lead to poor prognosis in HCC patients [69].
The protection of harmful pathogens depends on the activation of the immune system, which relies on the strict regulation of gene expression. There is compelling evidence that m6A modification is particularly critical in a variety of pathological and physiological immune responses, including T cell homeostasis and differentiation, inflammation, and type I interferon production [70]. In this study, it was found that HNRNPA2B1, HNRNPC, YTHDC1, METTL14, CBLL1, RBMX were significantly correlated with the level of immune cell infiltration in breast cancer, as well as with the Stromal Score, ESTIMATE Score, and immune Score. Through the TISIDB database, we found that the three m6A regulators had a close connection with Immuno inhibitors, Immuno stimulators, and MHC molecules in breast cancer. It is also suggested that the occurrence of breast cancer is related to the immune disorder caused by the abnormal expression of m6A.In addition, external factors, including Tumor-Infiltrating Lymphocytes (TIL), can lead to tumor resistance to immunotherapy, Tumor-Associated Macrophages (TAM), and Myeloid Suppressor Cells (MDSC). To deepen the understanding of the tumor immune microenvironment. For example, PD-L1 expression, TIL, TAMs, and MDSC play an increasingly important role. In this study, we assessed the association between m6A and levels of immune cell infiltration in TISIDB tumors. We also performed a bioinformatics analysis of m6A in breast cancer.
Several limitations should be noted in this study. First, TCGA data were used for survival analysis. Validation of mRNA and protein levels in surgical samples from breast cancer patients further supports the work of m6A modulators as viable clinical biomarkers. Finally, the function of the m6A regulatory factor in breast cancer will be further explored through functional loss analysis.
In conclusion, our study confirmed the dysregulation of tumor- associated m6A regulatory factor through bioinformatics analysis, which is associated with the prognosis of breast cancer patients and therefore can be used as a prognostic biomarker. In addition, we found that the expression of the m6A regulatory factor was associated with the level of immune invasion and the expression of immune regulatory factors in breast cancer. Our study suggests that m6A regulators may play a role as putative drug targets in breast cancer.
This study was approved by the Medical Ethics Committee of Cancer Hospital affiliated with Xinjiang Medical University. Our study was conducted according to the ethical standards of the Declaration of Helsinki.
All the authors approved the publication of the manuscript.
The authors thank the TCGA, GEO, and TISIDB databases for the availability of the data and data analysis of the xian tao academic website.
There is no conflict of interest.
This study was funded by Natural Science Foundation of Xinjiang Uygur Autonomous Region [Project No: 2021D01C410].
Yongtao Li and Muhairemu.Tuersuntuoheti designed this study, Yongtao Li and Muhairemu. Tuersuntuoheti Jianghua Ou, wrote and edited the manuscript, Xiaofang Chen and Xuelaiti. Paizula Lina Yi, reviewed and helped to analyze the data, and all authors read and approved the manuscript.
The authors thank the TCGA, GEO and TISIDB database for the availability of the data and data analysis of xian tao academic website.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
Citation: Tuersuntuoheti M, Chen X, Ou J, Yi L, Paizula X, Li Y (2023) Difference Expression of m6A Regulators in Breast Cancer and its Relationship with Immune Response. Chemo Open Access. 11:198.
Received: 25-Sep-2023, Manuscript No. CMT-23-27168; Editor assigned: 28-Sep-2023, Pre QC No. CMT-23-27168 (PQ); Reviewed: 12-Oct-2023, QC No. CMT-23-27168; Revised: 19-Oct-2023, Manuscript No. CMT-23-27168 (R); Published: 26-Oct-2023 , DOI: 10.35248/2167-7700.23.11.198
Copyright: © 2023 Tuersuntuoheti M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Sources of funding : This study was funded by Natural Science Foundation of Xinjiang Uygur Autonomous Region [Project No: 2021D01C410].