Journal of Clinical Toxicology
Open Access
ISSN: 2161-0495
ISSN: 2161-0495
Research Article - (2022)Volume 12, Issue 3
Formyl Peptide Receptors (FPRs) play an important role in the modulation of inflammation. There has however been a lot of ambiguity in the past with regard to the pro and anti-inflammatory profiles of FPR family members. A few FPR2/FPR1 dual agonists are reported in the public domain for their anti-inflammatory properties along with associated toxicities. Targeting FPR1 alone on the other hand has been shown to result into both positive and negative outcomes. We, therefore, aimed to develop FPR2 agonists which were selective against FPR1 followed by evaluating their potential in mitigating the non-resolving inflammation in asthma. Extensive Structure-Activity-Relationship (SAR) studies were conducted on the imidazole and benzimidazole chemotypes and a few molecules were shortlisted based on their in vitro profile and Absorption, Distribution, Metabolism and Excretion (ADME) properties. Molecules with acceptable biological profile were then further evaluated in the mouse models of asthma. In this manuscript, we are reporting the identification of RCI compounds with low nanomolar potency for FPR2 agonism and >10,000 fold selectivity over FPR1 in Ca2+ release assay. Interestingly, selective FPR2 agonists also showed potency in the mouse models of asthma. Our studies hereby confirm that FPR2 agonism alone is sufficient to address asthma and FPR1 could be spared to avoid the ambiguity and undesirable side effects.
Formyl Peptide Receptors (FPR); Mice; House Dust Mite (HDM); Leukocyte
Resolution of inflammation is an emerging concept which leads to the termination of inflammatory reaction by specific pro-resolving mediators [1,2]. By enabling healing and repair, the concept of resolution can be applied in treating various inflammatory diseases such as asthma, rheumatoid arthritis, alzheimer and cardiovascular diseases [3]. Formyl Peptide Receptors (FPRs) in this regard can be an important facilitator of this interesting phenomenon, as reported previously [4,5].
FPRs are G-protein-coupled receptors with three distinct family members; FPR1, FPR2, and FPR3 [6-11]. An increasing number of both peptide and non-peptide FPR1/2 dual agonists have been reported in literature with potent anti-inflammatory and immunological properties, none of these molecules however have progressed to the clinic [12-21]. We, therefore, did a careful evaluation of FPR1 and FPR2 specific knockout mice studies, reported in the literature and evaluated whether there can be a rationale to develop a selective agonist instead of a dual agonist to avoid any undesired response due to any one of them. We found that both FPR2 and FPR1 play an important role in the host defence as mice with loss of either FPR1 or FPR2 showed increased bacterial burden, neutrophil infiltration and a significant increase in the mortality compared with WT mice after a challenge with virulent E. coli clinical isolate or Listeria monocytogenes infections [22,23].
In inflammatory models, the two family members however, were found to show different outcomes. In a study using FPR1-/- mice, it has been found that FPR1 mediates neutrophil recruitment in the lung on endotoxin challenge leading to acute lung injury [24]. Role of Loss of FPR2 on one hand was found to result in unresolved LPS induced inflammation [25,26]. Two members have also been reported to play different roles in the progression of cancer. While FPR1 has been reported to be associated with various cancers [27], subcutaneously implanted LLC tumours in the FPR2 transgenic mice on the other hand, grew slower than those in the Wild-Type (WT) littermates [28].
These reports suggest that role of FPR1 in immunity and inflammation is very ambiguous. This has also been reviewed recently by Vacchelli, et al. [29]. We therefore proceeded with an understanding that activation of FPR2 alone could be a potential anti-inflammatory approach without compromising the innate immune response. Since no FPR2 selective agonists are reported in literature, we conducted a dedicated Structure Activity Relationship (SAR) studies in the imidazole and benzimidazole series to develop selective FPR2 agonists and evaluated them in moderate to severe models of asthma in mice to confirm whether activation of FPR2 alone is sufficient to combat asthma. All the molecules were evaluated in the cell based assays to confirm their mechanism of action and profiled for ADME studies prior to in vivo studies.
We report here with identification of RCI-1701306005, RCI- 1701309674, RCI-1701307669 as potent and selective agonists of FPR2 with nM potencies for FPR2 agonism and >10,000 fold selectivity over FPR1 in Ca2+ release assay using CHO cells overexpressing FPR2 and FPR1 receptors. RCI molecules also showed potent efficacy in four mouse models of moderate to severe asthma. Our findings suggest that agonism of FPR2 alone may be sufficient and FPR1 can be spared to achieve the desired pharmacological activity.
Cell culture and reagents
CHO cell lines overexpressing FPR2 and FPR1 receptors were generated in-house and propagated in Ham's F12 medium (Gibco Biosciences) supplemented with 10% (v/v) heat-inactivated Foetal Bovine Serum (FBS) (Lonza Group Ltd.) containing 100 μg streptomycin ml−1 and 100 U penicillin ml−1 in the presence of neomycin (400 μg/ml) (Calbiochem, EMD Chemicals). Cells were maintained at 37°C in a CO2 incubator with 5% (v/v) CO2. 15d-PGJ 2 EIA kit was obtained from Enzo Life Sciences Inc., Farmingdale, NY, USA. All other chemicals used in the study were purchased from Sigma-Aldrich Corporation. Stock solutions of all the test compounds were prepared in Dimethyl Sulfoxide (DMSO) and further dilutions were made in KHB buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4.7H2O, 1.2 mM KH2PO4, 4.2 mM NaHCO3, 11.7 mM D-Glucose, 1.3 mM CaCl2, and 10 mM HEPES) containing 30% DMSO. The final DMSO concentration in all the experiments was <0.5 %.
Experimental animals
All animal studies were conducted under the guidelines laid down by the Committee for Control and Supervision of Experiments on Animals (CPCSEA), Government of India and were approved by the Institutional Animal Ethics Committee (IAEC) of Daiichi Sankyo India Pharma Pvt. Ltd. 7 weeks old female BALB/c mice were used for all the studies and were procured from Vivo Bio Tech Ltd., Telangana, India and were housed in individually ventilated cages in experimental rooms. Mice had access to irradiated standard pellet feed and filtered autoclaved water provided ad libitum. In each experimental group, 6-8 animals were taken.
Transfection of CHO-K1 cells
CHO-K1 stable cell lines over-expressing Homo sapiens FPR2 full length (NCBI accession number NM_001462.3), Homo sapiens FPR1 full length, Mus musculus (NCBI accession number NM_008039.2), FPR1 (NCBI accession number NM_002029.3) were generated using pcDNA3.1 vector flanked by CMV immediate early promoter and BGH poly-A tail and neomycin as the selection marker. CHO-K1 cells were grown in 6 well plates in Ham's F12 medium (Gibco Biosciences) supplemented with 10% (v/v) heat-inactivated FBS (Lonza Group Ltd.) Containing 100 μg streptomycin ml−1 and 100 U penicillin ml−1 and 5 mM L-glutamine. One day before the transfection, media was changed to OptiMEM (Gibco). Transfection of plasmid DNA was done using lipofectamine 2000 (Life Technology) following the manufacturer’s protocol. 48 h after transfection, medium was replaced with fresh culture medium containing neomycin (400 μg/ml). Colonies were picked up after two weeks and analysed for the confirmation of FPR2/FPR1 expression by real-time PCR and western blot.
Ca2+ mobilization assay
The effect of test compounds on FPR2/FPR1 agonist-induced Ca2+ release was measured in this assay using a method established by us previously. Briefly, the CHO cells over-expressing human FPR2 or FPR1 receptor were harvested and suspended in KHB buffer containing 0.25 mM sulfinpyrazone and 3 μM Fura-2AM dye and incubated at 37°C in a CO2 incubator for 30 min, followed by 10 min incubation at room temperature. Cells were then washed thrice with KHB buffer containing 0.5% BSA and plated at a density of 5 × 104 cells/well in a 96-well black well clear bottom plate (Thermofisher).
To determine the agonistic activity, test compounds were added using the robotic arm fluorometric imaging system; Flex Station 3 Multi-Mode Microplate Reader (Molecular devices LLC, CA). Fluorescence intensity in each well was measured at an excitation wavelength of 340 and 380 nm and an emission wavelength of 510 nm every 5 s for 240 s at room temperature after automated addition of compounds. The maximum change in fluorescence during the first 3 m, over baseline was used to determine a response. The agonistic response induced by 5 nM of W peptide in the case of FPR2 and 5 nM of fMLF in the case of FPR1 was considered as 100% to calculate the % agonistic activity of the test compounds. The final DMSO concentration in all the wells was 0.5%. Doseresponse curves were generated using Prism 6 (GraphPad Software Inc., San Diego, CA, USA) and EC50 values were calculated using nonlinear regression analysis. Data was represented as an average of 3 independent experiments (n=3) with a % CV value <10 % CV and Z' value in the range of 0.9-1.
Erk1/2 phosphorylation and FPR2 expression studies
For phosphorylation studies, CHO-FPR2 cells were incubated with FPR2 agonists for the desired time point. Cells were harvested using 1x Trypsin EDTA, washed with ice-cold PBS and treated with ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton-X, 2.5 mM Sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na2VO4, 1 μg/ml Leupeptin and 1 mM PMSF. Lysed samples were centrifuged at 11,000 xg for 10 min at 4°C and supernatants were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and immuno-blotted with rabbit anti-human FPR2 or rabbit-anti- human P44/4 (Erk1/2) antibodies (Novus Biologicals). Blots were further rinsed with ice-cold PBS and probed further with goat- anti-rabbit IgG HRP (Cell Signaling Technology), washed further with ice cold PBS and analysed by the electrochemiluminescence (Millipore).
Total RNA extraction and RT-PCR
Total RNA from CHO-FPR2 overexpressing cells was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and purified using RNeasy Mini kits (Qiagen, Valencia, CA, USA). Total RNA (1–2 μg) was reverse-transcribed in a 25 μl reaction using Taqman reverse transcription reagents (Applied Biosystems, CA, USA). RT- PCR multiplex reaction was performed using gene-specific Taqman primers and probes (Applied Biosystems, CA, USA) and analysed using QuantStudio 6 Flex (Applied Biosystems, CA, USA) Real- Time PCR System. 18 s was used as an endogenous control. Relative Quantitation (RQ) of mRNA expression was analysed using QuantStudio™ Real-Time PCR Software v1.7.1 (Applied Biosystems, CA, USA).
15d-PGJ2 release in CHO-FPR2 cells
CHO-FPR2 cells were plated in a 96 well plate at a density of 5 × 103 cells per well. Cells were treated with desired concentrations of FPR2 agonists the next day for 16 h. 15d-PGJ2 levels in the cell culture supernatants of CHO-FPR2 cells were analysed using an enzyme immunoassay kit following the manufacturer’s instructions (Enzo Life Sciences Inc., Farmingdale, NY, USA). Plates were then read in a microplate reader at 405 nm and 15d-PGJ2 levels were calculated compared to untreated control.
Ovalbumin-induced airway eosinophilia
Female BALB/c mice weighing 25-30 g were sensitized with 2 mg of aluminium hydroxide in 100 μl normal saline and Ovalbumin (OVA) at 100 μg/mice, Intra-Peritoneal (I.P) from day 1 to day 14. Animals in the sham control group were administered 2 mg of aluminium hydroxide I.P.
Mice were challenged with OVA (50 μg/mice), dissolved in 50 μl normal saline under anaesthesia by intranasal (I.N.) route, followed by administration of vehicle (0.5 % methylcellulose) or test compound by the oral route on days 21 to 23. The sham-control group was challenged with 50 μg of ovalbumin (I.N.) dissolved in 50 μl of normal saline.
Mice were euthanized on day 23, with an overdose of sodium thiopentone administered (I.P.). Subsequently, Broncho-Alveolar Lavage Fluid (BALF) was collected by tracheostomy using ice-cold Hank's Balanced Salt Solution (HBSS). BALF was centrifuged at 2,000 xg for 10 min at 4°C and the cell pellet was re-suspended in 1 ml HBSS. The total leukocyte count in the cell suspension was enumerated by a veterinary haematology analyzer (Sysmex XT-2000iV, Sysmex, Japan). For obtaining differential leukocyte count, smears were prepared using 50 μl of the cell suspension on a frosted glass slide, stained with Leishman’s stain, and cell counts were taken by scientists who were blinded to the treatment groups.
House Dust Mite (HDM) induced chronic airway inflammation and airway hyperresponsiveness
Female BALB/c mice (25-30 g) in all the groups except the sham control group were instilled with Dermatophagoides pteronyssinus (HDM) extract (Greer Labs) (25 μg of protein in 20 μl normal saline), I.N.) Five days a week for 5 weeks. Sham-control animals received 20 μl of normal saline (I.N). Vehicle/test compounds/ reference standard were administered from week four till the last HDM exposure by the oral route (P.O.). 24 h after the last HDM instillation, mice were transferred to the body box of a whole-body plethysmograph (Buxco Laboratories). Subsequently, mice were exposed to the aerosolized saline following which the Penh was recorded for 5 min. Subsequently, the animals were sequentially exposed to aerosolized serotonin (Sigma-Aldrich) at 3, 10, and 20 mg/ml, and an increase in Penh was recorded for 10 min following each serotonin exposure. Airway Hyperresponsiveness (AHR) score was computed as the sum of the broncho-constrictor response to the aerosolized serotonin. Mice were euthanized 24 h after recording the AHR and Broncho-Alveolar Lavage Fluid was collected and processed for the enumeration of total and differential leukocyte counts as described in above section.
HDM and Complete Freund’s Adjuvant (CFA) induced airway inflammation
On day 0, animals in all the groups except the sham-control group were immunized subcutaneously on their back with 100 μl of an emulsion containing 50 μl of Complete Freund’s Adjuvant (Sigma-Aldrich) and 100 μg of Dermatophagoides pteronyssinus (HDM) extract (Greer Labs) dissolved in 50 μl of normal saline. Sham-control animals were administered an emulsion prepared with incomplete Freund’s adjuvant and normal saline. On day 14, animals in all the groups except the sham control group were intranasally challenged with 100 μg of HDM extract dissolved in 20 μl of normal saline. Sham-control animals were administered with normal saline (I.N.). 1 h before the HDM-challenge. Vehicle/test compounds/reference standard were administered (P.O.) on day 15 as well. The animals were euthanized 48 h after HDM-challenge; BALF was collected and processed for the quantification of total and differential leukocyte counts.
LPS induced Neutrophilia
Female BALB/c mice (25-30 g) were administered orally with the vehicle (0.5% methylcellulose) or test compound. One hour later, mice in all the groups except the normal control group were challenged with LPS (25 μg/mouse, I.N.). The normal control group was challenged with saline. The animals were euthanized 4 h after the LPS/saline challenge; BALF was collected and processed for the quantification of total and differential leukocyte counts.
Statistical analysis
At least n=3 or more experiments were conducted and all the experimental conditions were run in triplicate in cell-based assays. S.E.M. was calculated for each data point. Nonlinear regression analysis was used to determine the EC50 of the agonists using GraphPad Prism software. All animal experiments contained a minimum of 6-8 animals in each group. Significance of the data generated was confirmed by conducting Dunnet and student’s T test and a p-value of <0.05 were considered statistically significant.
RCI-1701306005, RCI-1701309674, RCI-1701307669 are potent and selective FPR2 agonists
RCI compounds were evaluated in the Ca2+ release assay using CHO cells over-expressing human and mouse FPR2. As a result of the extensive structure-activity relationship studies, RCI-1701306005, RCI-1701309674, RCI-1701307669 were identified (Figure 1) with nM potency in the Ca2+ release assay with no or little species variation using human and mouse FPR2 overexpressing cells and >10000 fold selectivity over hFPR1 (Figure 2, Table 1). RCI compounds did not show any liability against several other GPCRS such as muscarinic 2 (M2), muscarinic 3 (M3), Urotensin I, Urotensin II receptors and a panel of 42 kinases from various tyrosine and serine-threonine kinase families at 10 μM concentration.
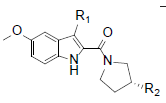 |
R1=H, | R1=Me, | R1=Cl, | R1=H, | R1=H, | R1=H, |
|---|---|---|---|---|---|---|
R2=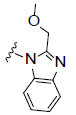 |
R2=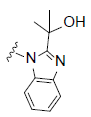 |
R2=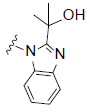 |
R2=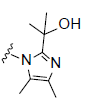 |
R2=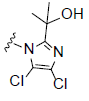 |
R2=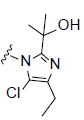 |
|
| RCI No. | 1701301195 | 1701306150 | 1701307669 | 1701306005 | 1701309674 | 1701301085 |
| hFPR2 Ca2+ EC50 (nM) | 5.74 ± 1.42 | 1.26 ± 0.67 | 4.01 ± 0.33 | 16.09 ± 2.32 | 3.48 ± 0.41 | 4.50 ± 1.22 |
| LogD | 2.7 | 3.3 | 3.6 | 2.1 | 2.9 | 3.2 |
| PAMPA | High | High | High | High | High | High |
| % PPB (m) | 97.34 ± 0.56 | 96.41 ± 0.49 | 99.12 ± 0.28 | 96.28 ± 0.67 | 99.30 ± 0.31 | 99.32 ± 0.22 |
| MS % remaining (m) | 48.24 ± 2.51 | 48.18 ± 3.13 | 67.41 ± 0.77 | 79.33 ± 0.25 | 75.56 ± 1.50 | 26.43 ± 3.24 |
Table 1: In vitro and ADME profile of selected NCEs (n=3).

Figure 1: Chemical structure of selected compounds.

Figure 2: Expression level of FPR1 and FPR2 in CHO cells overexpressing FPR1 and FPR2 respectively by real time PCR.
15d-PGJ2 release
FPR2 agonists have been previously reported to increase the release the levels of Cyclopentenone 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) which further bind and activate H-Ras leading to phosphorylation and activation of Erk1/2 [30-33]. We, therefore, tested the RCI compounds for 15d-PGJ2 release in CHO-FPR2 cells and found that the shortlisted FPR2 agonists led to an increase in the levels of 15d-PGJ2 in a concentration-dependent manner (Figure 3).

Figure 3: 15dPGJ2 release in CHO-FPR2 cells to demonstrate Resolution mediated activity by RCI compounds.
FPR2 agonists lead to the phosphorylation of Erk1/2 independent of PI3 kinase signalling cascade
Shortlisted FPR2 agonists; RCI-1701306005, RCI-1701309674, RCI-1701307669 led to the phosphorylation of Erk1/2 in FPR2- CHO cells, in a time-dependent manner, similar to FPR1/2 dual agonist, W peptide. Phosphorylation of Erk1/2 by RCI compounds was unaffected by LY294002 suggesting the PI3 kinase independent mode of action of these compounds for pro-survival signalling response (Figure 4).

Figure 4: Erk1/2 phosphorylation by FPR2 agonists in CHO-FPR2 cells, as analyzed by western blot and their relative densitometric values after normalization with beta actin (for FPR2 expression analysis) or total Erk1/2 for phosphor Erk1/2 expression analyses). a) Expression of FPR2 protein in CHO-hFPR2 cells, b) Phosphorylation of Erk1/2 by RCI-1701306005 and RCI-170130669 and c) W peptide and RCI-1701309674 in a time dependent manner.
Pharmacokinetic analysis
Pharmacokinetic (PK) analysis was conducted in mice to assess the exposure and bioavailability of these compounds and the selection of in vivo dose for efficacy studies in mice. Oral PK study was conducted at the dose of 10 and 100 mg while PK by intravenous (I.V.) route was done at 2 mg/kg. Blood samples were collected after 1, 2, 4, 8, and 24 h of administration of test compounds and several Pharmacokinetic (PK) parameters were analysed (n=3), (Figures 5 and 6, Tables 2 and 3). Data were presented as mean ± SEM. Significance of the data was confirmed by Dunnett and student’s T-test and P-value <0.05 was considered statistically significant [34].
| Test compound | Calcium release assay (EC50 in nM) | |||
|---|---|---|---|---|
| hFPR2-CHO | mFPR2-CHO | hFPR1-CHO | CHO WT | |
| RCI-1701306005 | 16.09 ± 2.32 | 42.15 ± 3.21 | >10000 | >10000 |
| RCI-1701309674 | 3.48 ± 0.41 | 3.34 ± 0.72 | >10000 | >10000 |
| RCI-1701307669 | 4.01 ± 0.33 | 2.52 ± 0.24 | >10000 | >10000 |
| W peptide | 0.56 ± 0.22 | 0.015 ± 0.01 | 1.27 ± 0.23 | >10000 |
Table 2: In vitro selectivity of shortlisted compounds in Ca2+release assays (n=3).
| Compound | Calcium release assay in hFPR2-CHO cells (EC50 in nM) |
|---|---|
| W peptide | 0.56 ± 0.2 |
| W peptide+WRW4 (10 µM) | >10000 |
| RCI-1701307669 | 4.01 ± 0.33 |
| RCI-1701307669+WRW4 (10 µM) | >10000 |
Table 3: Ca2+release assay in presence of FPR2 antagonist.

Figure 5: Chemotaxis assay in HL-60 cell line. a) Real time PCR assay to suggest expression of FPR2 in differentiated and un-differentiated HL-60 cells. b) %migration of differentiated HL-60 cells by RCI compounds.

Figure 6: Pharmacokinetic profile of RCI compounds by I.V (2 mg/kg) and P.O. (10 and 100 mg/kg) routes.
Efficacy of RCI compounds in mouse models of moderate to severe asthma
RCI compounds were evaluated in the animal models of asthma, representing progressively severe asthmatic conditions. In ovalbumin induced airway eosinophilia study using Balb/c mice, selective FPR2 agonists shortlisted by us, namely; RCI-1701306005, 1701309674 and 1701307669 demonstrated a dose-related inhibition of eosinophil counts with ID50 values of 11.8, 10.7 and 14.7 mg/kg, B.I.D. P.O respectively (Figure 7). In house dust mice (HDM) model also, RCI compounds showed significant inhibition of airway eosinophil influx at 10 and 30 mg/kg, P.O., B.I.D. as well as inhibition of airway hyper-responsiveness at 30 mg/kg, P.O., B.I.D. (Figure 8). In addition to the above two models, RCI compounds inhibited the levels of both eosinophils and neutrophil influx in the Broncho-Alveolar Lavage Fluid (BALF) of HDM+CFA treated mice as well, which represents a relatively more severe model of asthma (Figure 9). Finally, when tested in the LPS induced model of neutrophilia also, FPR2 agonists showed the inhibition of LPS induced increase in neutrophils (Figure 10). Dexamethasone (1 mg/kg, P.O., Q.D) was used as a method control in all the studies.

Figure 7: Dose response of RCI compounds in OVA induced airway eosinophilia mouse model in BALB/c mice to monitor change in eosinophil counts.

Figure 8: Evaluation of RCI compounds in HDM induced mouse model in BALB/c mice to monitor. a) Change in eosinophil counts by test compounds at 10 and 30 mg/kg P.O. dose to monitor change in eosinophil counts. b) Modulation of airway hyper responsiveness by RCI compounds at 30 mg/kg P.O.

Figure 9: Evaluation of RCI compounds in HDM+CFA-induced airway inflammation model in BALB/c mice at 10 mg/kg, P.O. to monitor. a) Change in eosinophil and, b) Neutrophil counts.

Figure 10: Evaluation of RCI compounds in LPS induced neutrophilia model in BALB/c mice at 30 mg/kg, P.O. to monitor the change in neutrophil counts as compared to vehicle control.
Asthma is a progressive disease and it has been reported that the disease severity is associated with impairment in the biosynthesis of pro-resolving mediators in these patients. We hypothesized that the pro-resolving properties of FPR2 may suit the chronic, non-resolving inflammatory conditions of the airways in asthma. FPR2/1 dual agonists although have shown promising resolution potential, none of these molecules have progressed to the clinic, till date. Selective agonists of FPR2 designed and synthesised by us were evaluated in mouse models of asthma to confirm whether activation of FPR2 is sufficient to retain the pro-resolution benefits of FPRs and FPR1 can be spared to overcome the FPR1 associated ambiguities.k their in vivo efficacy. In the OVA model, RCI- 1701306005, 1701309674, and 1701307669 compounds inhibited OVA-induced airway eosinophil influx in the Broncho-Alveolar Lavage Fluid (BALF) of mice, in
RCI-1701306005, RCI-1701309674, and RCI-1701307669 were shortlisted based on their in vitro cell based activity in FPR2 overexpressing CHO cells and ADME properties (Table 1). RCI molecules showed low nM potencies for FPR2 agonism and >10,000 fold selectivity over FPR1 in the Ca2+ release assay (Table 4). RCI compounds also demonstrated a time-dependent increase in the phosphorylation of extracellular signal-regulated kinase-1/2 (Erk1/2) in FPR2-CHO cells (Figure 3) in a PI3 kinase-independent manner and increased the levels of 15d-PGJ2 to mark their proresolution potential (Figure 4).
| Parameters | Cmax | AUC | F% | ||
|---|---|---|---|---|---|
| Dose (mg/kg, p.o) | 100 | 10 | 100 | 10 | 10 |
| RCI-1701306005 | 7.9 | 1.7 | 27.3 | 1.6 | 53 |
| RCI-1701309674 | 7.8 | 1.7 | 82.5 | 2.8 | 57 |
| RCI-1701307669 | 9.4 | 1.1 | 122.1 | 3.7 | 78 |
Table 4: In vivo Pharmaco-kinetic data of RCI compounds (n=2).
RCI compounds were evaluated in four distinct animal models of asthma to mark their in vivo efficacy. In the OVA model, RCI-1701306005, 1701309674, and 1701307669 compounds inhibited OVA-induced airway eosinophil influx in the Broncho-Alveolar Lavage Fluid (BALF) of mice, in a dose-dependent manner with ID50 values of 11.8, 10.7, and 14.7 mg/kg (BID, P.O.) respectively (Figure7). In the HDM model, RCI compounds showed significant inhibition of eosinophil influx in the BALF of mice at 10 and 30 mg/kg and a moderate inhibition of airway hyper-responsiveness at 30 mg/kg (Figure 8). RCI compounds also showed inhibition of eosinophil and neutrophil counts in the BALF of mice in HDM+CFA model (Figure 9). These compounds were also evaluated in the LPS induced neutrophilia model and showed potent inhibition of LPS induced neutrophil influx in the BALF. Potent in vivo efficacy demonstrated by RCI compounds in all the animal models of asthma evaluated by us confirms the antiasthmatic potential of FPR2 agonists. We, therefore, propose herewith that the development of selective FPR2 receptor agonists can be a potential approach for the management of moderate to severe asthma. Our studies also suggest that FPR2 agonism alone is sufficient to address asthma and we can spare FPR1 to avoid any ambiguity and undesirable pharmacological responses.
Resolution of inflammation is an important phenomena for the restoration of normal physiological state of a tissue after an inflammatory challenge. Endogenous Formyl Peptide Receptor 2 (FPR2/ALX) has been demonstrated to mimic such events and control inflammation as a result of their pro-resolving properties. Our study reports the identification of first-in-class therapeutic opportunity for asthma by the development of small molecule agonists of FPR2. These molecules were selective against its close family member FPR1 to avoid any toxicity concerns associated with previously reported FPR2/1 dual agonists without compromising their efficacy in mouse models of moderate to severe asthma.
We acknowledge Dr. Ashok Patra, Director, Imgenex India for discussions in the generation of FPR2 and FPR1 stable cell lines.
The author declared no conflict of interest.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Visaga SA, Kalonia H, Verma V, Sinha S, Singh SK, Upadhyay S, et al. (2022) Developing Selective FPR2 Agonists can be a Potential Approach to Treat Moderate to Severe Asthma? J Clin Toxicol. 12:510.
Received: 13-May-2022, Manuscript No. JCT-22-17280 ; Editor assigned: 16-May-2022, Pre QC No. JCT-22-17280 (PQ); Reviewed: 30-May-2022, QC No. JCT-22-17280 ; Revised: 06-Jun-2022, Manuscript No. JCT-22-17280 (R); Published: 13-Jun-2022 , DOI: 10.35248/2161-0495.22.12.510
Copyright: © 2022 Visaga SA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.