Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 0, Issue 0
Blockade of gap junctions (GJs) has been shown to reduce seizures in different epilepsy models. Gap junction mediated, electrically-coupled neuronal networks have been implicated in neuronal synchronization, which is the hallmark of seizure activity. To further understand the role of GJs in seizures, in particular hippocampal seizures, we evaluated electrophysiological responses in the dentate gyrus subfield of the hippocampus, including GABA-mediated recurrent inhibition, seizuregenic stimulation, and seizure activity in response to perforant path kindling in Cx36 knockout (KO) mice compared to wild-type (WT) controls. In anesthetized mice, Cx36 KO mice were characterized by enhanced GABA-mediated recurrent inhibition. Stimulation of the perforant path at 10 Hz for 10 sec (i.e., 100 pulses) markedly enhanced population spike (PS) amplitudes and reduced paired-pulse GABA-mediated inhibition during the stimulation, induced an after discharge for approximately 10 sec at 7-10 sec into the stimulation, and suppressed PS amplitudes postictally for 5-10 min in both KO and WT mice. Repeated epochs of 10 Hz for 10 sec stimulation of the perforant path at 20 min intervals resulted in progressive and persistent disinhibition of paired-pulse responses in WT, but not KO mice. In freely-behaving seizure studies, stimulation of the perforant path (10 Hz for 10 sec) once/day resulted in progressive Stage I-IV seizures in both WT and KO mice. Once Stage IV was achieved, another Stage IV seizure could be elicited each day with the same behavioral response (i.e., Stage V). The threshold for kindled seizures was significantly greater in KO mice compared to WT controls for most stages of seizures. The Cx36 antagonist mefloquine (MFQ) and the typical anticonvulsant pentobarbital reduced Stage IV seizures in both WT and KO mice. Knock-out mice were more sensitive to the anti-epileptic effects of the typical anti-convulsant pentobarbital (20 mg/ kg). Taken together, these findings support the emerging view that reduction in GJ-mediated GABA electrical coupling reduces seizures, perhaps through enhancement of GABAergic feedback inhibition, which results from the uncoupling of the GABA recurrent interneurons from the resistive load that is inherent in their electrical connectivity.
<Seizures results from uncontrolled, synchronous cortical activity [1], which is thought to be caused by synaptic hyperexcitability [2]. Excessive excitatory glutamate release or reduced inhibitory GABA release has been implicated in hyperexcitability and uncontrolled neuronal activity, resulting in seizures and ultimately epilepsy. Epilepsy is characterized by spontaneous, recurrent seizures. When the balance of inhibition-excitation is disrupted, epileptiform seizures develop [3,4]. GABA release plays a vital role in maintaining a sensitive balance of excitation-inhibition. GABA receptor antagonists have been found to yield epileptogenic seizures [5] and enhanced GABA receptor-mediated auto-inhibition can prevent hyperexcitability [6].
Animal models have proven to be effective in studying epilepsy in humans [7]. The kindling model is a physiologically-relevant model of epileptogenesis [8]. Kindling is accomplished through repeated electrical stimulation to discrete structures in the brain [9], which leads to a state of persistent hyperexcitability [10]. The first stimulation typically evokes a local after discharge, which is of relatively short duration and elicits minimal behavioral effects. However, with repeated stimulations, seizures progress in duration, severity and distribution [11,12], transitioning from focal, non-overt seizures to generalized motor seizures [10]. Kindling not only results in generalization of seizure activity, but also results in a permanent state of excitability due to plasticity, or complex reorganization of circuitry [11]. Kindling facilitates determinations regarding site of action [8], and allows for the unambiguous study of anticonvulsant drugs [13], as no other drugs are present in the brain to complicate interpretations regarding specific drugs effects, as is often the case with chemically-induced seizures. Such is the case with stimulation of the perforant path, the primary excitatory neocortical input to the dentate gyrus of the hippocampus [14]. It is well-known that repeated stimulation of the perforant path for several days induces sub-clinical, and ultimately, full-clinical seizures. In this kindling model of epilepsy-induction, current is administered directly to the perforant path, allowing neural circuitry to behave independently of drug or other physiological elements. The dentate gyrus is susceptible to seizures because of its intrinsic circuitry that allows oscillatory feedback excitation. Presumably, when GABA inhibition is compromised, feedback-excitation via recurrent excitatory interneurons becomes open-loop, resulting in an oscillation of synaptic and neuronal activity. These after discharges have shown to elicit spreading of uncontrolled neuronal firing to other areas of the CNS with subsequent stimulations to the perforant path.
Because of their inherent ability to rapidly connect and synchronize networks of neurons, gap junction (GJs) have stimulated a lot of recent interest in epilepsy research. Gap junctions are pores constructed between cells from approximately 20 members of the connexin (Cx) protein family by aggregation of six Cx proteins, forming a connexon which associates with a connexon on a neighboring cell [15]. These pores allow for the coupling of membrane voltages between neighboring neurons [16], thereby providing a means for intercellular communication in both the developing and mature nervous system [17]. Many different connexin proteins are expressed in the various brain cell-types, including glial cells [15]. Glia are now increasingly linked to seizure generation as they are extensively connected via GJs [Cx30, 43, and 26; [18]]. Two types of Cxs are ubiquitously expressed in mammalian neurons: Cx36, found mostly in GABA interneurons of the mature CNS [19,20]; and Cx45, expressed throughout the nervous system during development, but less so in the adult [21]. The ability of GJs between neurons to rapidly connect neurons [16] has linked them to the propagation of high-frequency oscillations, rhythmicity, neuronal synchrony, and epileptic seizures [18]. They often govern oscillations in GABA neuron networks [22-24], potentially leading to increased excitatory activity or decreased inhibitory activity and thus promoting epileptogenesis [16]. Pro-convulsant drug studies have suggested a role for GJ-mediated intercellular communication in ictogenesis [15]. Despite the fact that GJs are involved in neuronal oscillations and synchrony, evidence for their role in behavioral models has lagged behind the molecular and physiological evidence. The GJs between GABA-releasing interneurons in the mature brain of mammals are of the Cx36 variety, which are found in the hippocampus and other areas of the brain [25-28]. Epilepsy has been correlated with changes in Cx expression and intercellular coupling [29]. However in most cases, these experiments with Cx expression and epilepsy were performed with already epileptic subjects [30-33]. Indeed, many studies have shown that GJs play a role in epilepsy, as seizures can be ameliorated by GJ blockers and exacerbated by GJ openers [31,34-40]. For example, mice lacking neuron-specific Cx36 GJs evince reduced kainate-induced [41] and 4-aminopyridine-induced seizures [42]. One study showed that Cx36 knock-out (KO) mice appear to be more susceptible to druginduced seizures using the excitotoxin kainic acid [15].
The hippocampus is a well-known focal point for seizure initiation and ultimately propagation to the neocortex [43]. The dentate gyrus subfield of the hippocampus is particularly susceptible to seizures, and appears to be the primary locus of temporal lobe epilepsy. The vulnerability of the dentate gyrus to seizures may result from disrupted GABAergic synaptic transmission as the pathological changes that occur in the dentate during epilepsy include loss of GABAergic inhibitory interneurons [44-46] and sprouting of axons [47-49]. Synchronization, characteristic of epileptiform bursts, has been recorded in the hippocampus, and has been theorized to be mediated by electrical coupling via GJs [50]. Cx36 GJs are found in hippocampal neurons [51]. Indeed, we have previously shown that Cx36 GJ expression is co-localized with GAD in GABA neurons in the ventral tegmental area (VTA) as well as other CNS structures including the hippocampus [52]. Regarding physiological evidence for GJs in the hippocampus, after discharges, as well as spontaneous bursts of hippocampal population spikes, which are representative of synchronized action potentials, persist even when chemical synaptic transmission is blocked [50]. Given the speed of synchronization, it was suggested that ionic changes in the extracellular space were not likely to be responsible for synchronizing neurons in the hippocampus [50]. Further evidence for the role of hippocampal GJs in epileptogenesis has also been provided through dye/tracer coupling studies [35]. Specifically, parvalbumin (PV)-containing GABAergic interneurons of the hippocampus have been suggested to have GJs, which likely facilitate synchronization of oscillatory neuronal networks [53,54]. We have also evaluated the role of Cx36 GJs in modulating ventral tegmental area (VTA) dopamine and GABA neuron excitability [25,55,56] as well as ethanol effects on these neurons [56,57]. It was in these studies that we noted that Cx36 KO mice did not exhibit ethanol withdrawal-induced seizures compared to WT mice (unpublished observation). Thus, the purpose of this study was to better evaluate the role of Cx36 GJs in perforant path kindling, a well-known, nonpharmacological method to study seizure activity. We evaluated dentate evoked field potentials, GABA receptor-mediated recurrent inhibition, and seizuregenic perforant path stimulation in anesthetized WT vs Cx36 KO mice, as well as GABA and GJ pharmacology on perforant path kindled seizures in freely-behaving WT vs Cx36 KO mice.
Animal subjects
The care and use of mice and experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Brigham Young University which met or exceeded NIH guidelines. Only adult male mice (PND 60-120) were used in this study. Connexin-36 (Cx36) knock-out (KO) mice were originally created on a C57/Bl6 background in the laboratory of Dr. David Paul (Harvard Medical School, Cambridge, MA). In Cx36 KO mice, the Cx36 coding sequence was replaced by a LacZ-IRES-PLAP reporter cassette [58]. Activation of the Cx36 promoter results in expression of the cytoplasmic protein β-gal, and cells that would normally generate Cx36 transcripts expressed the β-gal reporter throughout their cytoplasmic domain. Cx36 KO founders were a gift of Dr. Marla Feller (University of California, San Diego, CA), and were compared against age-matched C57/Bl6 wild-type (WT) mice (Charles River, Wilmington, MA). Once weaned at PND 21, all mice were housed in groups of four/cage and placed on a reverse light/dark cycle with lights ON from 8 PM to 8 AM. Mice were given ad libitum access to solid food and water.
Extracellular recordings and data acquisition
Evoked field-potentials and hippocampal EEG were recorded in isoflurane-anesthetized mice by single 3.0 M NaCl filled micropipettes (1-3 MΩ; 1-2 microns i.d.) stereotaxically oriented into the dentate gyrus [coordinates (in mm from bregma): 2.0 AP; 2.0 L; 1.4-1.8 V from dura] with a piezoelectric microdrive (EXFO Burleigh 8200 controller and Inchworm, Victor, NY). Extracellular potentials were amplified with an Axon Instruments Multiclamp 700A amplifier and displayed on a Tektronix (Beaverton, OR) digital oscilloscope. Hippocampal electroencephalographic (EEG) and evoked field potentials were amplified at 5,000X and 100X and filtered at 0.1-50 Hz and 0.1 Hz - 10 kHz kHz (-3dB), respectively. Extracellular potentials were digitized with National Instruments (Austin, TX) NB-MIO-16 data acquisition boards on Macintosh computers at 200 Hz for hippocampal EEG activity (for monitoring after discharges) and 20 kHz for evoked responses and single-unit activity (at 12-bit resolution).
Stimulation
Square-wave pulses (50-1000 μA; 0.15 ms duration; average frequency=0.05 Hz) were generated by a constant current isolation unit triggered by a MASTER-8 pulse generator or by computer. Field-potentials were elicited in the dentate gyrus by stimulation of the perforant path (for recordings in dentate gyrus) with insulated, bipolar stainless steel (130 μm) electrodes located in the angular bundle [coordinates (in mm from bregma): 3.6 AP; 2.8 L; 2.0-2.5 V]. Stimulation of the perforant path elicited population EPSPs (pEPSPs) when recorded from the stratum moleculare of the dentate. Population EPSP slopes were recorded in the dendritic fields of the dentate and measured between 10-90% of the peak amplitude of the initial falling phase of the waveform. Recordings made from the cellular levels of the dentate yielded a population spike (PS) superimposed on the positive-polarity pEPSP/pIPSP complex. Fine adjustments were made in dorso-ventral positioning of recording and stimulating electrodes to maximize PS amplitudes, which were measured by a median filter and peak detection algorithm. An input/output analysis was performed on three trials each to determine the current intensity needed to achieve threshold and maximum PS amplitudes. The intensity was then adjusted to a level that produced 50% maximum PS amplitudes. Pairedpulse curves and 10 Hz stimulation studies were performed at the 50% maximum stimulus level and averaged across three trials at each interstimulus interval. Paired-pulse curves were generated by testing various intervals (0.010-10.0 s) of orthodromic paired stimuli.
Surgical procedures and post-operative care
Mice were anesthetized with isoflurane and secured in a stereotaxic frame. Body temperature was maintained at 37.4°C by a heating pad driven by a feedback-regulated controller. Under asceptic conditions, bipolar, insulated stainless-steel electrodes were stereotaxically oriented into the diagonal band of the perforant path through a 1 mm cranial trephined hole. In addition, two 1 mm holes were drilled and stainless steel screws were tapped into the holes in order to stabilize the implant. The probes were then cemented in place with dental acrylic. Isoflurane anesthesia was maintained at 2% during the surgical procedure. The analgesic buprenorphine was injected 10 min before discontinuing anesthesia. Analgesic therapy was continued every 12 hours for a minimum of 48 hours, as needed for the control of pain. One WT and one Cx36 KO mouse were placed in each cage and housed in a temperature and humidity-controlled environment with a 12 h. reverse light/dark cycle. Food and water were available ad libitum. Mice were given at least one week of recovery following surgical implantation before behavioral testing. The health status of each mouse was examined at least once daily.
Kindling procedures
We implanted 18 mice with stainless-steel, insulated bipolar stimulating electrodes through 1 mm cranial trephined holes and into the perforant path located 3.6 mm posterior, 2.8 mm lateral, and 2.5 mm ventral, in reference to Bregma. In addition, 2 additional 1 mm holes were drilled and screws tapped into the holes in order to stabilize the implant. The probes were cemented to the screws and skull with dental acrylic. When the animals woke up from surgery, we followed strict post-operative care as described below in order to ensure positive well-being of the mice.
After 7 days post-surgery, each mouse was placed in a plexiglass, open-field seizure chamber with piezoelectric transducers cemented to the underside of the suspended floor of each chamber that measured motor activity and tremors during seizures. We allowed the mice to acclimate in the seizure chamber for 5 minutes pre-stimulation. While in the seizure chamber, we elicited after discharges with 150 μsec biphasic pulses at 10 Hz for 10 sec at 0.5 mA (an epoch of stimulation) in awake, freely-behaving mice and recorded their seizure activity both by observation of seizure stages and movement on the piezoelectric pressure board. They were then monitored for 10 min after the stimulation in the chamber. With one stimulation epoch (100 pulses), an after discharge is typically produced in the hippocampus, but is restricted to the hippocampus. However, when this stimulation paradigm (100 pulses to the perforant path) is repeated once a day, every day, the excitation becomes progressively amplified in intensity and spreads out of the hippocampus and into other areas of the CNS. This is termed perforant path kindling [11,59]. We continued stimulation once each day, kindling the mice, until each mouse reached stage 4 seizures. The seizure stages were: Stage 1=Wet dog shakes, mouth or facial movements; Stage 2=Chewing motion, head bobbing, or twitching; Stage 3=Unilateral or bilateral forelimb clonus; Stage 4=Rearing and falling or tonic/clonic seizures; Stage 5=Four consecutive Stage 4 seizures. In six WT and six KO mice demonstrating Stage 5 seizures, we administered an intraperitoneal injection of the Cx36 GJ blocker mefloquine (MFQ) each day for 4 days one hour before stimulation. We then waited one week and re-established Stage 5 seizures. Finally, we administered an intraperitoneal injection of the GABAA receptor agonist pentobarbital each day for 4 days one hour before stimulation.
Drug preparation and delivery
Pentobarbital and mefloquine hydrochloride were obtained from Sigma Chemical and dissolved in physiologic saline and injected intraperitoneally.
Analysis of responses
Analog waveforms and stimulation events were processed with National Instruments (Austin, TX) LabVIEW virtual instrument software and Wavemetrics IGOR Pro software (Lake Oswego, OR) on PC-type computers. Three trials were averaged at each stimulus level of the stimulus/response curves and for each interval of the paired-pulse curves. Results for control and drug treatment groups were derived from calculations performed on evoked field potential and paired-pulse data and are expressed as means ± S.E.M. Differences between WT vs KO for paired-pulse responses, response to 10 Hz stimulation and seizure scores were compared with one-way analysis of variance (ANOVA). The criterion of significance was set at p<0.05. Analysis software included Statistical Analysis Software (SAS Institute, Inc), Microsoft Excel, and Igor Pro (Wavemetrics, Oswego, OR). Significance levels are indicated on graphs with asterisks and hashtags and correspond to significance levels P<0.05, 0.01 and 0.001. Figures were constructed with Igor Pro software.
Evoked field potentials and paired-pulse responses in the dentate of wild-type and connexin-36 knock-out mice
Stimulation of the perforant path in anesthetized mice at 0.1 Hz elicited a negative-going population EPSP (pEPSP) recorded in the dendritic layer (stratum moleculare) and a negative-going population spike (PS) recorded in the cellular layer (stratum granulosum) that was superimposed on the positive-going pEPSP, whose slope and amplitude were dependent on stimulus level: threshold, 50% maximum (Figures 1A and B). Equipotent paired-stimulation of the perforant path generated biphasic test/conditioning response curves in the dentate of WT mice, as we have previously reported [60-62]. Paired-pulse responses in the dentate were characterized by complete inhibition of the test PS amplitude at interstimulus intervals (ISIs) below 40 msec and potentiation between 40-2060 msec (Figure 1A; truncated at 80 msec). Paired-pulse responses in Cx36 KO mice were characterized by prolonged inhibition compared to WT mice (Figure 1B). There was a significant difference in paired-pulse inhibition at 40 msec ISI between WT and KO mice (Figure 1C; P =0.002, F(1,11)=33.6; n=6 each).
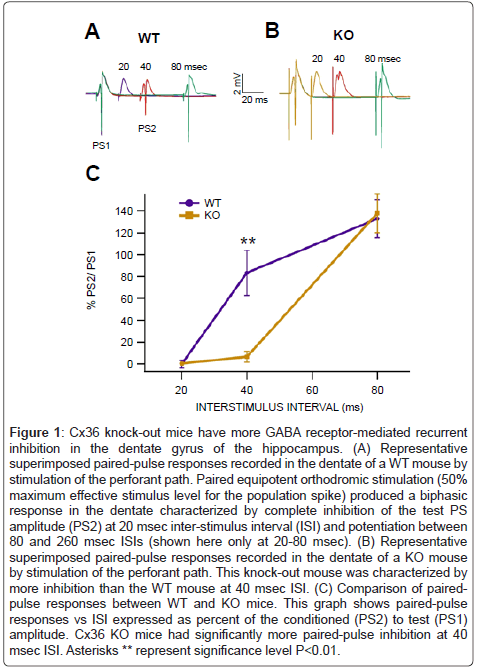
Figure 1: Cx36 knock-out mice have more GABA receptor-mediated recurrent inhibition in the dentate gyrus of the hippocampus. (A) Representative superimposed paired-pulse responses recorded in the dentate of a WT mouse by stimulation of the perforant path. Paired equipotent orthodromic stimulation (50% maximum effective stimulus level for the population spike) produced a biphasic response in the dentate characterized by complete inhibition of the test PS amplitude (PS2) at 20 msec inter-stimulus interval (ISI) and potentiation between 80 and 260 msec ISIs (shown here only at 20-80 msec). (B) Representative superimposed paired-pulse responses recorded in the dentate of a KO mouse by stimulation of the perforant path. This knock-out mouse was characterized by more inhibition than the WT mouse at 40 msec ISI. (C) Comparison of pairedpulse responses between WT and KO mice. This graph shows paired-pulse responses vs ISI expressed as percent of the conditioned (PS2) to test (PS1) amplitude. Cx36 KO mice had significantly more paired-pulse inhibition at 40 msec ISI. Asterisks ** represent significance level P<0.01.
Transient loss of GABA receptor-mediated recurrent inhibition in the dentate with single 10 Hz stimulation: relative resistance in connexin-36 knock-out mice
Stimulation of the perforant path at 10 Hz for 10 sec in anesthetized mice elicited a complex oscillation of PS amplitudes during and 5-10 min after the stimulation epoch in both WT and KO mice. An afterdischarge measured with the same electrode, but filtered from 0.1-100 Hz, was also generated around 7-8 sec into the stimulation epoch and lasted for approximately 10 sec after the stimulation. Paired stimulation (20 msec ISI) of the perforant path at 10 Hz for 10 sec elicited nearly identical responses as that produced by single stimulation (i.e., PS amplitudes and afterdischarges) but facilitated the monitoring of GABA receptormediated recurrent inhibition (Figure 2). During the stimulation epoch, responses in WT mice were characterized by marked enhancement of PS amplitudes during the first 5 sec, followed by disinhibition of paired-pulse responses from 5-8 sec, and suppression of PSs at 8-10 sec (Figure 2A). After the stimulation, PSs recovered in about 5-10 min. The recovery phase following stimulation was also accompanied by disinhibition of the conditioned PS at 20 msec which lasted 3-5 min. In KO mice, 10 Hz stimulation elicited a similar profile as in WT mice with the same marked enhancement of PSs, followed by depression and disinhibition (Figure 2B). However, there was significantly more pairedpulse disinhibition in WT compared to KO mice (P=0.05, F(1,11)=5.0; n=6 each; Figure 2C).
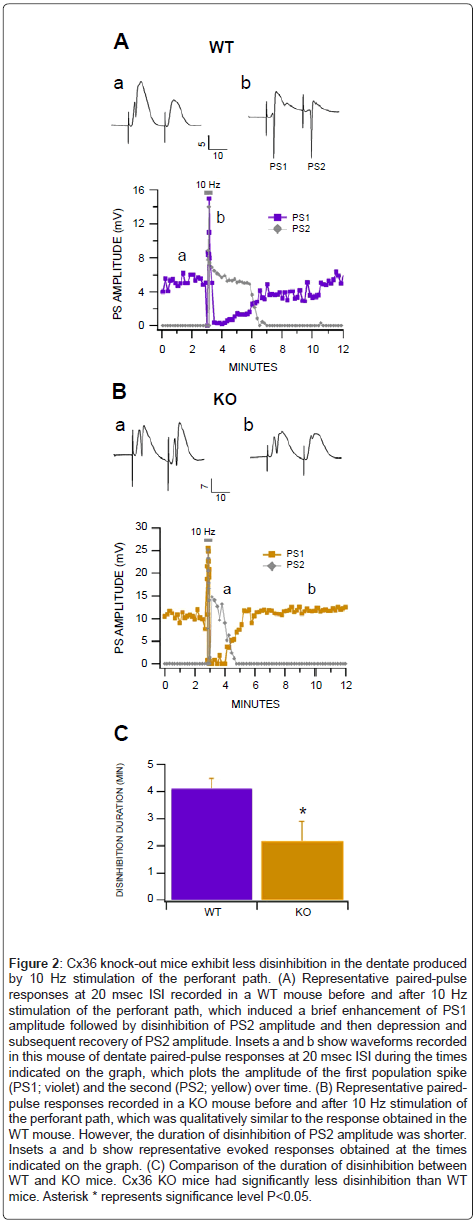
Figure 2: Cx36 knock-out mice exhibit less disinhibition in the dentate produced by 10 Hz stimulation of the perforant path. (A) Representative paired-pulse responses at 20 msec ISI recorded in a WT mouse before and after 10 Hz stimulation of the perforant path, which induced a brief enhancement of PS1 amplitude followed by disinhibition of PS2 amplitude and then depression and subsequent recovery of PS2 amplitude. Insets a and b show waveforms recorded in this mouse of dentate paired-pulse responses at 20 msec ISI during the times indicated on the graph, which plots the amplitude of the first population spike (PS1; violet) and the second (PS2; yellow) over time. (B) Representative pairedpulse responses recorded in a KO mouse before and after 10 Hz stimulation of the perforant path, which was qualitatively similar to the response obtained in the WT mouse. However, the duration of disinhibition of PS2 amplitude was shorter. Insets a and b show representative evoked responses obtained at the times indicated on the graph. (C) Comparison of the duration of disinhibition between WT and KO mice. Cx36 KO mice had significantly less disinhibition than WT mice. Asterisk * represents significance level P<0.05.
Persistent loss of GABA receptor-mediated GABA inhibition in the dentate with repeated epochs of 10 Hz stimulation: relative resistance in connexin-36 knock-out mice
It is well-known that seizures can be induced by repetitive stimulation of the perforant path. As there was a small but significant difference between responses in WT vs KO mice to a single 10 Hz stimulation to the perforant path (Figure 2), we evaluated the effects of repeated 10 Hz stimulation epochs on dentate PSs and GABA receptor-mediated recurrent inhibition in WT vs KO anesthetized mice. As recovery from a single 10 Hz stimulation epoch appeared to be complete in 10 min we repeated 10 Hz stimulation epochs every 20 min. We compared the response of the 1st to the 10th 10 Hz epoch as well as baseline responses in WT vs KO mice. In WT mice, although baseline conditioning PS amplitudes did not change significantly (P>0.05), the conditioned PS amplitude recorded at the 20 msec ISI became progressively disinhibited (Figure 3A). The 10th 10 Hz stimulation epoch induced its typical transient enhancement of PS amplitudes and suppression of PS amplitudes after the stimulus; however, the conditioned PS amplitude remained persistently disinhibited. In KO mice, 10 Hz stimulation induced its typical response with no persistent disinhibition (Figure 3B). Figure 3C compares paired-pulse responses in WT vs KO mice for the 1st and 10th 10 Hz stimulation epochs. There was a significant and persistent loss of GABA inhibition (20 and 40 msec ISIs) in WT mice compared to KO mice (P=0.004, F(1,15)=11.4; n=4 each; Figure 3C).
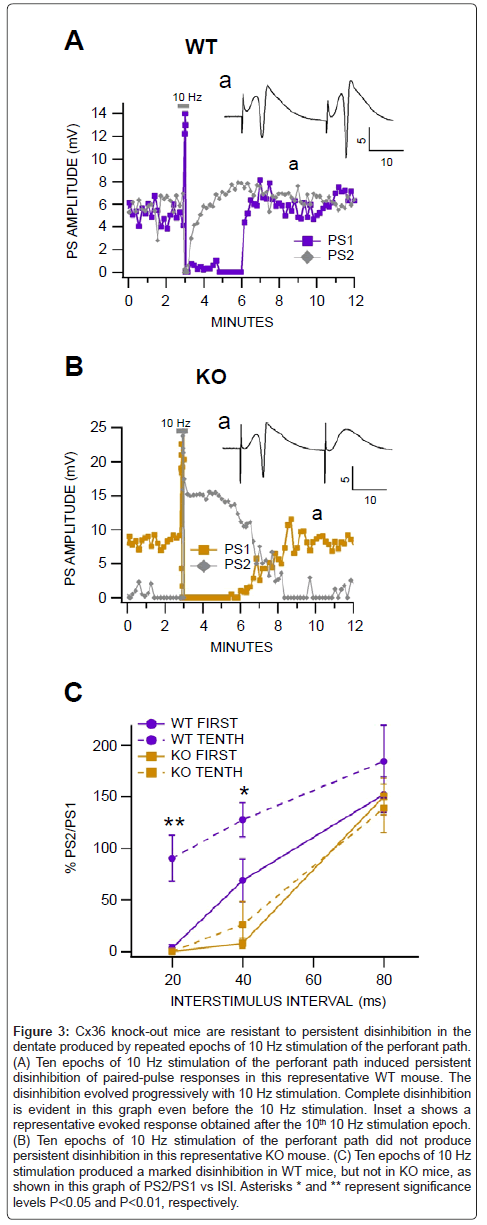
Figure 3: Cx36 knock-out mice are resistant to persistent disinhibition in the dentate produced by repeated epochs of 10 Hz stimulation of the perforant path. (A) Ten epochs of 10 Hz stimulation of the perforant path induced persistent disinhibition of paired-pulse responses in this representative WT mouse. The disinhibition evolved progressively with 10 Hz stimulation. Complete disinhibition is evident in this graph even before the 10 Hz stimulation. Inset a shows a representative evoked response obtained after the 10th 10 Hz stimulation epoch. (B) Ten epochs of 10 Hz stimulation of the perforant path did not produce persistent disinhibition in this representative KO mouse. (C) Ten epochs of 10 Hz stimulation produced a marked disinhibition in WT mice, but not in KO mice, as shown in this graph of PS2/PS1 vs ISI. Asterisks * and ** represent significance levels P<0.05 and P<0.01, respectively.
Kindled seizures: relative resistance in connexin-36 knockout mice
Perforant path kindled seizures were evaluated in 8 WT and 8 KO freely-behaving mice that were implanted chronically with cranial electrodes and stimulated once at 10 Hz for 10 sec each day. As one KO mouse died prematurely, we included in our analysis data from 8 WT and 7 KO mice. The day each mouse reached Stage 1-4 seizure was recorded. There were significant differences in seizure threshold between WT and KO mice (Figure 4A). Cx36 KO mice had significantly higher thresholds for perforant path kindled seizures at nearly all stages (Stage 1: P=0.02, F(1,14)=6.4; Stage 2: P=0.43, F(1,13)=0.66; Stage 3: P=0.04, F(1,13)=5.3; Stage 4: P=0.005, F(1,13)=11.8). .
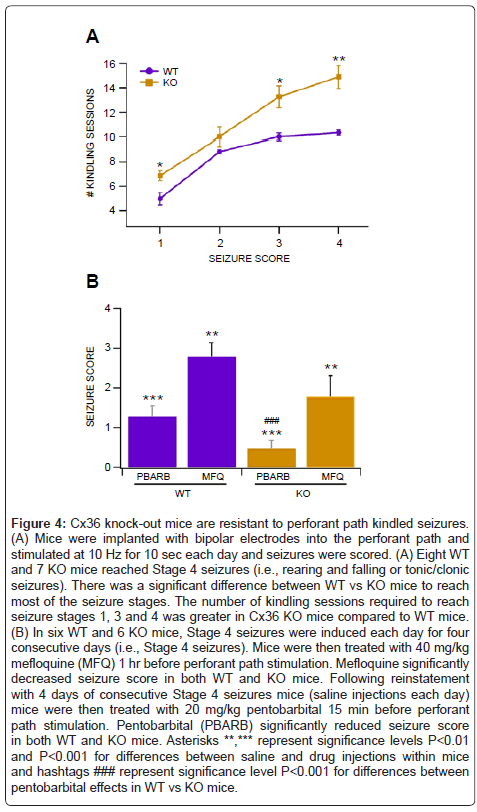
Figure 4: Cx36 knock-out mice are resistant to perforant path kindled seizures. (A) Mice were implanted with bipolar electrodes into the perforant path and stimulated at 10 Hz for 10 sec each day and seizures were scored. (A) Eight WT and 7 KO mice reached Stage 4 seizures (i.e., rearing and falling or tonic/clonic seizures). There was a significant difference between WT vs KO mice to reach most of the seizure stages. The number of kindling sessions required to reach seizure stages 1, 3 and 4 was greater in Cx36 KO mice compared to WT mice. (B) In six WT and 6 KO mice, Stage 4 seizures were induced each day for four consecutive days (i.e., Stage 4 seizures). Mice were then treated with 40 mg/kg mefloquine (MFQ) 1 hr before perforant path stimulation. Mefloquine significantly decreased seizure score in both WT and KO mice. Following reinstatement with 4 days of consecutive Stage 4 seizures mice (saline injections each day) mice were then treated with 20 mg/kg pentobarbital 15 min before perforant path stimulation. Pentobarbital (PBARB) significantly reduced seizure score in both WT and KO mice. Asterisks **,*** represent significance levels P<0.01 and P<0.001 for differences between saline and drug injections within mice and hashtags ### represent significance level P<0.001 for differences between pentobarbital effects in WT vs KO mice.
Pharmacology of kindled seizures in wild-type vs connexin-36 knock-out mice
In order to evaluate the role of GJs in perforant path kindled seizures, we tested the effects of the the Cx36 antagonist mefloquine (MFQ). We have previously demonstrated that MFQ blocks electrical coupling between VTA GABA neurons in adult mice [25,55] and reduces brain stimulation reward [52]. While MFQ is selective for GJs, it has other physiologically-relevant non-specific effects. However, in our previous studies, we determined that MFQ’s GJ blocking effects were optimal at 1 hr after administration. Thus, we tested the effects of MFQ on kindled seizures 1 hr after administration. Six WT and 6 KO mice reached criterion for Stage 5 seizures (4 consecutive days of Stage 4 seizures). We administered 40 mg/kg MFQ to each mouse 1 hr before 10 Hz for 10 sec stimulation of the perforant path on 4 successive days and averaged the seizure scores over the 4 days (Figure 4B). Intraperitoneal injection of 40 mg/kg MFQ significantly reduced seizures in both WT and KO mice (WT: P=0.005, F(1,11)=12.8; KO: P=0.002, F(1,11)=18.5). There were no significant differences in MFQ effects between WT vs KO mice (P = 0.13, F(1,11) = 2.7). The following day after pentobarbital treatment, mice were injected with saline 15 min before stimulation of the perforant path. Stage IV seizures were restored in all mice for 4 consecutive days (i.e., Stage V). Intraperitoneal injection of the anticonvulsant pentobarbital (20 mg/kg) significantly reduced seizures in both WT and KO mice (WT: P=1.2E-06, F(1,11)=106; KO: P=7.0E-09, F(1,11)=314). Cx36 KO mice were more sensitive to pentobarbital than WT mice (P = 0.001, F(1,11)=21.2).
Connexin-36 KO mice were characterized by significantly more paired-pulse inhibition that WT mice. Hippocampal recurrent inhibition is presumably mediated by GABAergic interneurons interpolated in feedback loops to hippocampal principal cell types [2,4,5,14]. We, and others, have previously demonstrated, in the rodent dentate gyrus, that paired-pulse inhibition can be dramatically reduced by the GABAA antagonist bicuculline [39], by conditioning stimuli applied to the medial septum [38], during theta activity produced by tail-pinch (unpublished observation), during REM sleep [36], or by sensory activation [22]. One mechanism that might explain the enhanced recurrent inhibition in the dentate of Cx36 KO mice is that the lack of electrical coupling between GABAergic interneurons in the dentate, in particular basket cells that presumably mediate feedback inhibition, might allow for more inhibition due to the uncoupling of GABA neurons from their network load. In otherwords, neurons that are connected via GJs typically have lower rates of activity due to loading of other cells in the network. If basket cells were chronically hyper excitable due to this uncoupling one might expect to observe enhanced recurrent inhibition.
A single 10 Hz for 10 sec stimulation of the perforant path elicited after discharges, marked changes in dentate field potential responses and paired-pulse inhibition, both during and after the stimulation. There are a plethora of studies demonstrating that all the subfields of the hippocampus undergo long-term potentiation (LTP) or depression (LTD) depending upon the frequency and duration of stimulation. In general, brief, high frequency stimulation evokes LTP in the dentate gyrus, while low frequency stimulation evokes LTD. Previous protocols have exhibited that 10 Hz stimulation in the hippocampus successfully elicits behavioral seizures [63] and also exhibit prolonged afterdischarges [64]. This method has been shown to effectively model seizures in animal models [64] and lead to self-sustaining status epilepticus [65-67]. On the other hand, high frequency electrical stimulation was unable to induce fully kindled seizures [67]. Thus, low frequency stimulation around frequencies associated with theta rhythm (i.e., 5-10 Hz) is effective in evoking epileptic seizures that result in lasting changes in the dentate gyrus. Other than what we demonstrate here, others have shown that GABA inhibition in the dentate gyrus is lost with perforant path stimulation [68], possibly though interfering with the normal function of GABAA receptors [68,69] and modifying receptor binding [70]. This implies that epileptic seizures could be a result of decreased inhibition to the dentate gyrus. By monitoring paired-pulse inhibition during and after a single 10 Hz stimulation we demonstrate compelling physiological evidence that GABAergic recurrent inhibition is transiently diminished in the dentate gyrus, which may be an harbinger for persistent loss of GABA inhibition seen with perforant path kindling. Many in vitro experiments have failed to show epileptiform responses or loss of GABA inhibition with stimulation frequencies similar to that used in vivo. In fact, epileptiform bursts rarely occur in the hippocampal slice preparation, even in epileptic animals, with similar stimulation paradigms unless GABA inhibition is severely compromised. However, Shao and Dudek [71] have recently found that repetitive stimulation of the perforant path at physiological frequencies in the range of theta rhythm can induce epileptiform bursts in the dentate of hippocampal slices in rats with kainite-induced epilepsy. Regardless, we show here that a single 10 Hz for 10 sec stimulation results in disinhibition of paired-pulse responses and that repeated epochs of 10 Hz stimulation produces progressive and persistent disinhibition of paired-pulse responses in the dentate gyrus of naive WT mice, suggesting that the seizures induced in freely-behaving animals by perforant path kindling evolve from loss of GABAergic recurrent inhibition. Interestingly, we show that Cx36 KO mice are relatively resistant to this loss of recurrent inhibition. It is plausible that this resistance is simply the result of enhanced GABA inhibition as a result of uncoupling of GABA neurons from their GJ-mediated resistive load. As GJ coupling allows neurons to all fire together, GJs load neurons so they do not fire spontaneously. Rather, they fire synchronously and less frequently than those that are not linked through GJs. Thus, in Cx36 KO mice, the GABA-releasing interneurons may fire spontaneously and out of synchrony, as they do not load together. It appears that these interneurons fire more frequently, as we see more inhibition and less seizure susceptibility in KO mice.
In WT freely-behaving mice, 10 Hz for 10 sec stimulation of the perforant path induced overt manifestations of seizure activity (i.e., Stage 1) which progressed to full tonic-clonic seizures in 9-10 days. This is consistent with what others have reported in rodents with the perforant path kindling procedure. Most importantly, Cx36 KO mice were relatively resistant to kindled seizures. This finding raises questions about the role of Cx36 gap junctions in ameliorating seizures, contrasting what has been originally speculated in studies with druginduced seizures [15]. Perhaps the hyper-inhibitory characteristics of Cx36 KO GABAergic interneurons are unable to override the excitatory state produced by drug-induced seizures. One advantage of the kindling model of seizure induction is that it facilitates the evaluation of neural circuitry in the absence of drugs. Kindling not only resulted in generalization of seizure activity, but appears to result in a permanent state of excitability due to a natural process of GABA disinhibition, or complex reorganization of circuitry [11]. However, perforant path kindling, while a valuable paradigm for studying hyperexcitability and GABAergic synaptic transmission in the absence of drugs, does not fully model epilepsy, as there are little or no spontaneous seizures.
The Cx36 blocker MFQ, as well as the typical anti-convulsant pentobarbital, significantly reduced Stage 4 seizures in WT mice. Pentobarbital was more effective at reducing seizures in KO mice. In some ways, this is not surprising, since GABA recurrent inhibition appears to be stronger in KO than WT mice, as shown by our pairedpulse studies. This might be explained by selective activation of different populations of GABA interneurons in the dentate that inhibit each other, some of which may express Cx36 GJs while others do not. This remains to be evaluated in a future study. However, MFQ’s ability to reduced seizures in Cx36 KO mice was most unexpected. While MFQ is selective for Cx36 GJs [72], it is not specific to GJ effects only. Indeed, studying Cx36 GJs pharmacologically is complicated by the lack of drugs that act specifically and exclusively on GJs. Mefloquine is the most promising [for review [73]]. Besides being used clinically as an antimalarial drug, MFQ disrupts intracellular calcium [74-76], acts as an antagonist at serotonin 5HT3 receptors [77] and also as an acetylcholinesterase inhibitor [78]. Thus, MFQ has multiple effects that need to be considered with any interpretation regarding its effects. However, we have shown previously in midbrain slice studies that its GJ blocking effects predominate, and may be exclusively operational, after 1 hr of administration [55], which was the time delay we chose to study its effects on kindled seizures. Furthermore, in this previous study, we demonstrated that GABA inhibition to VTA dopamine neurons following MFQ treatment was markedly enhanced, nearly 5X over control or baseline conditions, suggesting that uncoupling GABA neurons from their electrotonic load increases their rate of firing and ultimately their inhibitory drive to dopamine neurons. Gap junctions can be modeled as variable resistors that effect cellular and network physiology in both open and closed conformations. With this view of GJ physiology, a possible explanation for the data presented here is that GABA interneurons in the dentate are no longer coupled to the resistive load of a GABA network and thus fire more autonomously. Whether GJ-mediated interneuron synchrony facilitates faster firing, slower firing, or both, depending on where the GJs are located and whether their currents are timed with transmitter release [22,24,79], it appears from this study that lacking GJs or blocking GJs leads to greater release of GABA onto target neurons, in this case dentate granule cells. This might explain why Cx36 KO mice evinced more paired-pulse inhibition in the dentate and were relatively resistance to the disinhibition produced by 10 Hz stimulation and to perforant path kindling.
In summary, by using electrophysiological and behavioral measurements associated with stimulation of the perforant path, we found that Cx36 KO mice had more dentate GABAergic recurrent inhibition, had less disinhibition with single 10 Hz stimulation, lacked persistent disinhibition with repeated 10 Hz stimulation, and had a lower threshold for perforant path kindled seizures compared to WT mice. In addition, the Cx36 antagonist MFQ lowered seizure score and the anti-convulsant pentobarbital was more effective in KO than WT mice. Taken together, these findings implicate Cx36 GJs in perforant path kindling and suggest that KO mice are protected from seizures due to uncoupling of GJ-coupled GABA neurons and enhanced GABA neurotransmission in the dentate gyrus subfield of the hippocampus. This study presents the view of GJs as being more akin to variable resistors that effect cellular and network physiology in both open and closed conformations. With this view of GJ physiology, a possible explanation for the data presented here is that when MFQ blocks GJs, the electrically-coupled VTA GABA network becomes uncoupled, releasing each cell from the resistive network load. The result being that individual GABA neuron activity is no longer synchronized with its neighboring GABA neurons, but rather is released from the resistive load of the network and thus enabled to fire more autonomously. But whether GJ-mediated GABA interneuron synchrony facilitates faster firing, slower firing, or both depending on where the GJs are located and whether their currents are timed with transmitter release [22,24,79], it appears from this study that lacking or blocking Cx36 GJs leads to greater release of GABA onto dentate granule cells. The findings of this study strengthen the argument that GJs serve as critical modulators of brain activity and may serve as strategic targets for the clinical treatment of epilepsy.
Samuel I Shin and Scott C Steffensen conceived and designed the experiments; Samuel I Shin, Dallin J Andersen, Micah Hansen D, Jordan T Yorgason and Scott C Steffensen performed the experiments; Samuel I Shin and Scott C Steffensen analyzed the data:; Samuel I Shin, Nathan D Schilaty, David D Busath, and Scott C Steffensen wrote the paper.