Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2013) Volume 2, Issue 2
Natural oligopeptides comprise of 2-50 amino acid residues and present in virtually all living organisms.
Despite the fact that several functions of these peptides are known, the exact nature of mechanistic activity need to be identified through dissecting conformational and structural details. In this study, we performed conformational analysis using molecular dynamics (MD) simulation in order to study the dynamic behavior of three tripeptides: L,LGly- Phe-Phe (commercial), L,L-Phe-Gly-Phe, and L,L-Phe-Phe-Gly in explicit water model.
Keywords: Tripeptide, Molecular dynamics, Conformational analysis, AMBER
Natural oligopeptides consist of 2-50 amino acid residues [1,2]. These oligopeptides are generally considered as ‘regulatory’ due to their involvement in multiple regulatory mechanisms and physiological processes including nervous, endocrine, and immune [3] specific events. The oligopeptide toxins of eukaryotes play an important role in the regulation of interspecie relationships. Similarly, antimicrobial oligopeptides of prokaryotes regulate competition in occupation of ecological niches and act as signal molecules in intercellular communications [4].
Comprehensive knowledge of an oligopeptides specific chemical structure is based on the determination of its amino acid sequence, detection of modifications of its N and C terminals, Post Translational Modifications (PTMs), and S-S or other internal chemical bonds lying in distant amino acid residues including the additional peptide bond produced upon cyclization. Due to the lack of natural content (10-15- 10-12 M) [5], determination of accurate 2D structure for oligopeptides is an arduous task. Moreover, their cellular localization is also an unsolved problem associated with isolation. Therefore, highly sensitive biochemical and immunohistochemical techniques are required for their analysis.
In recent years, computational approaches have been increasingly developed to explore the interactions of various biological macromolecules at structural level. Among them, Molecular Dynamics (MD) simulation enables the exploration of biological systems at atomic level and potentially provides valuable insights about their dynamic properties. Thus, the study of time dependent behavior make MD a powerful technique for understanding the intricacies of the interactions which govern the folding and binding processes of biological macromolecules.
In this work, we carried out extensive MD simulation studies for three oligopeptides: L,L-Gly-Phe-Phe (commercial), L,L-Phe- Gly-Phe, and L,L-Phe-Phe-Gly (synthesized by Pr S. GHALEM) [6]. These oligopeptides were simulated for 10 ns in explicit water model at 300 K using the ff99SB force field. The main objective of this study is to understand the structural stability of mentioned oligopeptides containing uncoordinated aromatic side chains. Generally, amino acids bearing an aromatic side chain, such as tyrosine and phenylalanine play important role in the complex formation with metal ions. These side chains do not serve as a site of ligand; however, their presence largely influences the complexation by various effects. For example, steric effect, the π-d effect and hydrophobic effect [7,8] are the main influencing agents.
Indeed, a pentapeptide such as enkephalin (Tyr-Gly-Gly-Phe- Met or Tyr-Gly-Gly-phe-Leu) of low molecular weight [9] has been reported [10-12] as an endogenous ligand and acts as antagonist to the opioid receptors. Any change of the Tyrosyl receptor residues at the N-terminus may inhibit the biological activity [13]. The relative spatial disposition of aromatic side chains of phenylalanine is also interesting due to a possible resemblance of enkephalin. Complexation of copper (II) by [5-Leucine]- and [5-Methionine]- enkephalin has been studied under physiological conditions [14]. To specify the biologically active conformation of enkephalin [15-16], it was observed that in the sequence of enkephalin, the modification of residues L-Tyr1, Gly2, and the L-Phe4 causes a significant decrease in activity. On the contrary, the residue Gly2 could be replaced by another acid of D conformation [17] without significant change in activity.
The atomic labelling and the torsion angles of interest are indicated in figure 1. The φ (phi, involving the backbone atoms C’-N-Cα-C’), ψ (psi, involving the backbone atoms N-Cα-C’-N) and ω (omega, involving the backbone atoms Cα-C’-N- Cα) are the backbone dihedral angles of protein. Thus, φ controls the C’-C’ distance, ψ controls the N-N distance and ω controls the Cα-Cα distance.
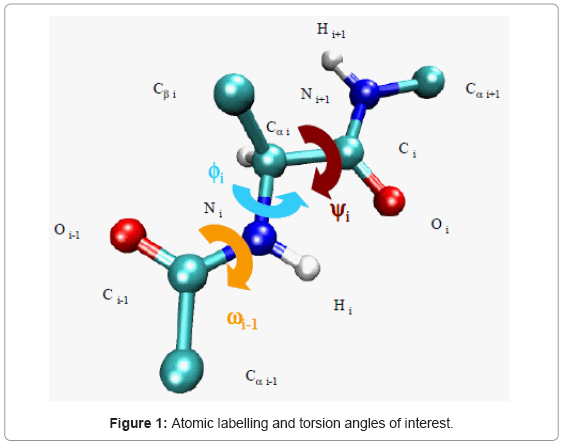
Figure 1: Atomic labelling and torsion angles of interest.
MD simulations
The MD simulations were carried out using AMBER10 [18] at a temperature of 300 K. This temperature was chosen because our primary objective was to study the dynamics of the oligopeptides at physiologically relevant conditions. The starting coordinates of the oligopeptides were constructed with the amino acids database included in Hyperchem 7.5. The simulations were carried out using an explicit solvent environment with AMBER package version 10. The solvated system for the oligopeptides molecule was prepared using an explicit water box in the Xleap module of AMBER10 by a periodic box of TIP3PBOX 8 waters. The solvent box dimension was identical for all three oligopeptides: 18.77×18.77×18.77 Å3. The number of water molecules was 906 identical for these tripeptides.
Since these coordinates are not optimized, a first minimization by molecular mechanics (1,000 steps of steepest descent followed by 1,000 steps of conjugate gradient minimization) was made with AMBER10 to get rid of any unfavorable contacts and then equilibrated for 120 ps to bring the temperature from ~0 K to 300 K, after which production dynamics were run for 10 ns. Force field ff99SB was used for these oligopeptides. All simulations were run with the SANDER module of AMBER with SHAKE algorithm [19] (tolerance=0.0005?) to constrain covalent bonds (involving hydrogens), using periodic boundary conditions. The further parameters were as follows: a 2 fs time step, a temperature coupling [20] step, a 8 ? cuttoff was applied to the Lennard-Jones interaction, and constant pressure of 1 atm. The periodic boundary conditions were used to negate the surface effects at the box boundaries. The Particle Mesh Ewald (PME) summation method [21] was used to calculate the electrostatic potential. The bonds containing hydrogen atoms were constrained using SHAKE [22] algorithm. As a result, bond interactions involving hydrogen atoms were not calculated concomitantly. This preliminary study was aimed at finding the most energetically favorable structure for all three cases, which was found at 3696.2 ps for L,L -Gly-Phe-Phe, at 3855.6 ps for L,L-Phe-Gly-Phe, and at 5634.8 ps for L,L-Phe -Phe-Gly. These conformations were found with a “home-made” Perl script that merely checked all energy values, finding the lowest one and giving the associated structure [23].
The PTRAJ module of AMBER10 was used to analyze and process the trajectory and coordinate files from SANDER, where the analyses included carrying out superimposition, clustering analysis of hydrogen bonds, calculating fluctuations in bonds, angles or dihedrals, correlation functions etc. and the Root Mean-Square Deviations (RMSDs) studies.
Finally, isoenergy contour maps were generated with ORIGIN and SURFER 9 [24]. The exact positions of the minima in these maps were subsequently calculated by minimizing the energy without restraints of those grid points indicating minima on the maps.
The calculated adiabatic conformational energy surfaces (φ, ψ) for the three tripeptides were presented as total energy contour maps in figures 2-4, (the lowest energy conformers for each compound are shown). The contours are separated by 1 kcal/mol and ranged from 1 to 10kcal/mol. The energies are given relative to the lowest minimum. Geometries and relative potential energies of the predicted minima are listed in tables 1-3, and the lowest energy conformers for each compound are shown in figures 2- 4.
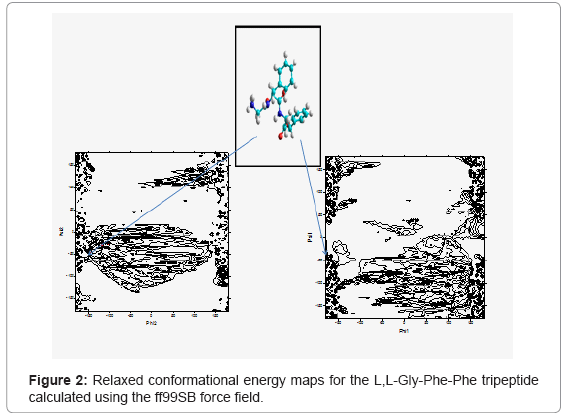
Figure 2: Relaxed conformational energy maps for the L,L-Gly-Phe-Phe tripeptide calculated using the ff99SB force field.
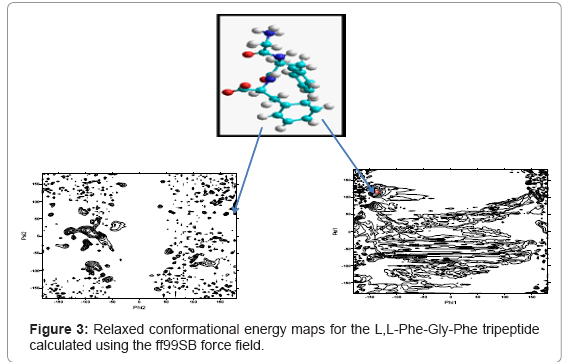
Figure 3: Relaxed conformational energy maps for the L,L-Phe-Gly-Phe tripeptide calculated using the ff99SB force field.
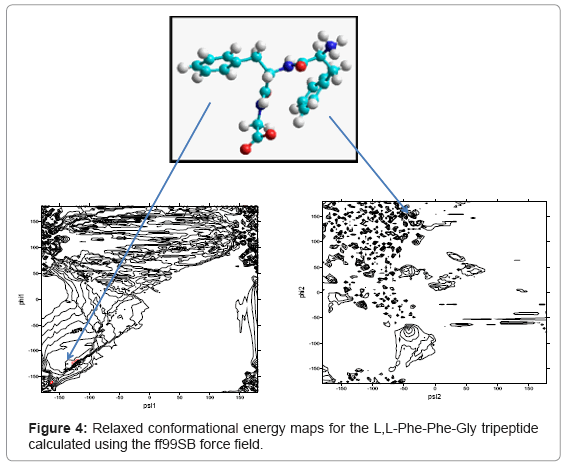
Figure 4: Relaxed conformational energy maps for the L,L-Phe-Phe-Gly tripeptide calculated using the ff99SB force field.
| TE | φ1 | φ2 | ψ1 | ψ2 | Omega1 | Omega2 | |
|---|---|---|---|---|---|---|---|
| Conformer1 | -4975.023 | -175.34 | 174.31 | 115.98 | -10.54 | -102.95 | 88.56 |
| Conformer2 | -4974.554 | -161.67 | 160.58 | 147.10 | 4.089 | 129.69 | -91.56 |
| Conformer3 | -4969.982 | 179.44 | 177.60 | 126.45 | 160.75 | -104.77 | 149.71 |
Table 1: Total energy, torsional angles for the conformers with low energies of the L,L-Gly- Phe-Phe tripeptide.
| TE | φ1 | φ2 | ψ1 | ψ2 | Omega1 | Omega2 | |
|---|---|---|---|---|---|---|---|
| Conformer1 | -6168.924 | -177.17 | -109.38 | 138.19 | 164.62 | -109.38 | 145.41 |
| Conformer2 | -6162.356 | -168.48 | -73.69 | 120.77 | 158.49 | -73.69 | 132.42 |
| Conformer3 | -6159.94 | -169.81 | -86.44 | 126.26 | 161.015 | -86.44 | -134.4 |
Table 2: Total energy, torsional angles for the conformers with low energies of the L,L-Phe- Gly-Phe tripeptide.
| TE | φ1 | φ2 | ψ1 | ψ2 | Omega1 | Omega2 | |
|---|---|---|---|---|---|---|---|
| Conformer1 | -4405.32 | 179.31 | -122.99 | 157.36 | 124.24 | -122.99 | 166.16 |
| Conformer2 | -4397.93 | 166.41 | -92.50 | 129.74 | 162.51 | -92.50 | 68.31 |
| Conformer3 | -4386.12 | -170.36 | -68.01 | 155.03 | 154.73 | -68.01 | 74.37 |
Table 3: Total energy, torsional angles for the conformers with low energies of the L,L-Phe- Phe-Gly tripeptide.
System root-mean-square displacement
RMSD is an important parameter for describing the system stability. Figures 5-7 demonstrate the function curves between mass weighted RMSD fit and the time of L,L-Gly-Phe-Phe, L,L-Phe-Gly-Phe, and L,LPhe- Phe-Gly, during dynamics simulation process, respectively.
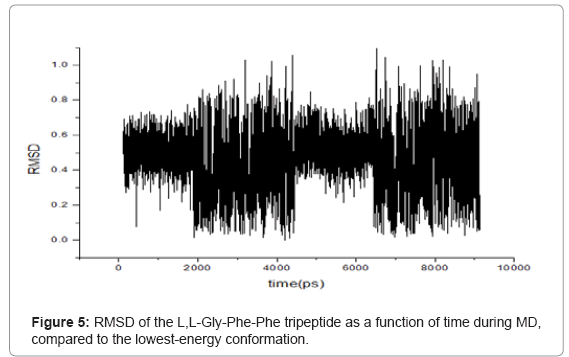
Figure 5: RMSD of the L,L-Gly-Phe-Phe tripeptide as a function of time during MD, compared to the lowest-energy conformation.
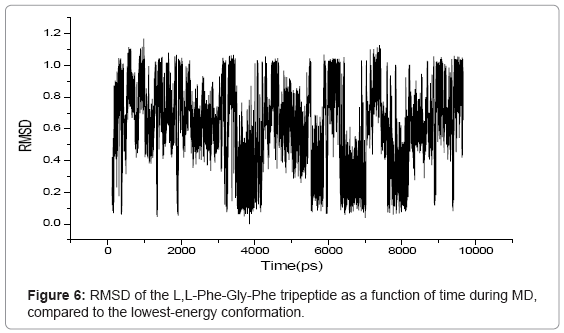
Figure 6: RMSD of the L,L-Phe-Gly-Phe tripeptide as a function of time during MD, compared to the lowest-energy conformation.
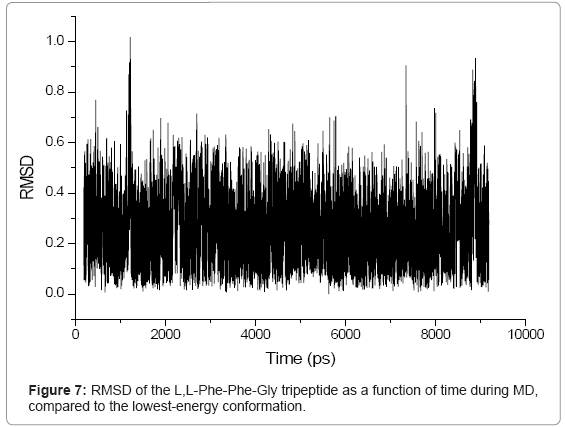
Figure 7: RMSD of the L,L-Phe-Phe-Gly tripeptide as a function of time during MD, compared to the lowest-energy conformation.
Computation of Phi/Psi for modeled oligopeptide
In the present study, structure prediction of oligopeptides has been made through the torsion angles phi (φ)/psi (ψ). The combination of φ and ψ angles fully determines the conformation of an oligopepide. Figures 2- 4 show the distribution of sterically allowed and energy minimized conformations in φ/ψ peptidic space obtained from our explicit solvent MD simulations for 10 ns in case of the three tripeptides L,L-Gly-Phe-Phe, L,L -Phe-Gly-Phe, and L,L-Phe-Phe-Gly, respectively. These figures also show the best conformers (which have the low energies). The conformers and their corresponding φ/ψ angular values are reported in tables 1-3 for the L,L-Gly-Phe-Phe, L,L-Phe-Gly- Phe, and L,L-Phe-Phe-Gly, respectively.
The monitoring of kinetic, global, and potential energies along with the trajectory, as well as the pressure and temperature indicate that the global periodic system is stable and does not present simulation artifacts. During MD simulations, kinetic energy increased slowly, while potential energy decreased gradually. The total energy of a system remains conserved during simulation, whereas the temperature fluctuation is caused by the inter-convention of the kinetic and potential energy components [25].
The RMS variations as a function of time were displayed in figures 5- 7 for the L,L-Gly-Phe-Phe, L,L-Phe-Gly-Phe, and L,L-Phe-Phe- Gly, respectively. These data indicated that all three systems were equilibrated after 500 ps. The visual examination of 10 ns trajectories for all three oligopeptides indicated a stable behavior of peptides during simulation. To evaluate more closely, an RMSD analysis was performed. The calculated RMSD difference between the 3D structures at the start of simulations (constructed with the amino acids databases included in Hyperchem 7.5), and the low-energy conformers including only heavy atoms is 0.19 ?, 0.41 ? and 0.1 ? for the L,L-Gly-Phe-Phe, L,L-Phe-Gly-Phe, and L,L -Phe-Phe-Gly, respectively. This shows that all three oligopetides are quite stable.
During the simulation of oligopeptides, we noticed that for L,LGly- Phe-Phe, the most stable conformer was obtained at 3696.2 ps, while for L,L -Phe-Gly -Phe and L,L-Phe-Phe-Gly peptides, the stable conformers were found at 3855.6 ps and 5634.8 ps, respectively.. This indicates that L,L-Gly-Phe-Phe peptide is less flexible than the L,L-Phe- Gly-Phe, which is less flexible than L,L-Phe-Phe-Gly peptide due to the steric effect.
Clearly, these three tripeptides, which differ only in the position of the Glycine residue, as obtained by calculations: EL,L -Phe-Gly -Phe < EL,L- Gly-Phe-Phe < EL,L-Phe-Phe-Gly, exhibited the order of stability as follows: L,LPhe- Gly-Phe>L,L-Gly-Phe-Phe>L,L-Phe-Phe-Gly, which is due to the steric (volume and position of the phenyl group) and electronic (electronic distribution) effects.
To confirm this, we made a calculation of electron population at DFT level with the 6-31G* basis using Gaussian 09 program. For the L,L-Phe-Gly-Phe tri peptide, we noted that the positions of phenyl groups were homogeneous (Figure 8) resulting in steric and electronic equilibrium. In contrast, for the other tripeptides L,L -Gly-Phe-Phe and L,L-Phe -Phe-Gly, the these positions of phenyl groups were not homogeneous. Moreover, we observed a significant increase in the stability upon orientation of aromatic group in C-terminal position (L,L-Gly-Phe-Phe) compared to the tripeptide containing the same amino acids, while the phenyl residue remained into N-terminal (L,LPhe- Phe-Gly) orientation. . Thus, for the L,L-Gly-Phe-Phe tripeptide, the two phenyl groups are located in the same side (Figure 9), while for the L,L-Phe -Phe-Gly tripeptide, the phenyl group of the 3rd amino acid is located in the same side as the N-terminus (Figure 10), which create an electronic repulsion, resulting in the instability of this tripeptide compared to the others.
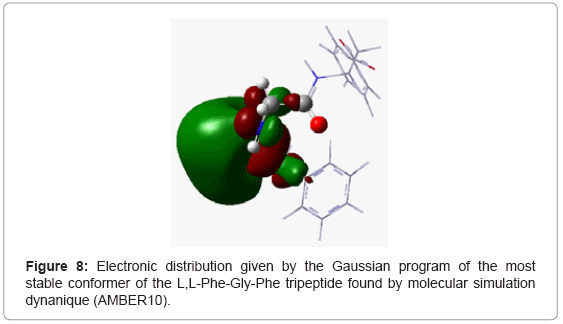
Figure 8: Electronic distribution given by the Gaussian program of the most stable conformer of the L,L-Phe-Gly-Phe tripeptide found by molecular simulation dynanique (AMBER10).
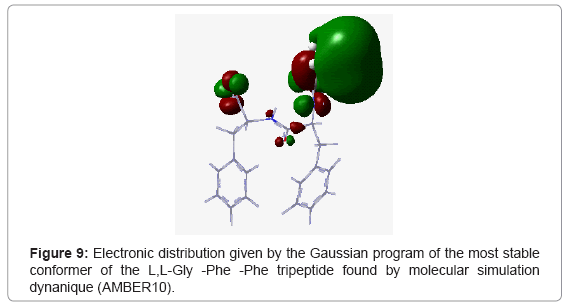
Figure 9: Electronic distribution given by the Gaussian program of the most stable conformer of the L,L-Gly -Phe -Phe tripeptide found by molecular simulation dynanique (AMBER10).
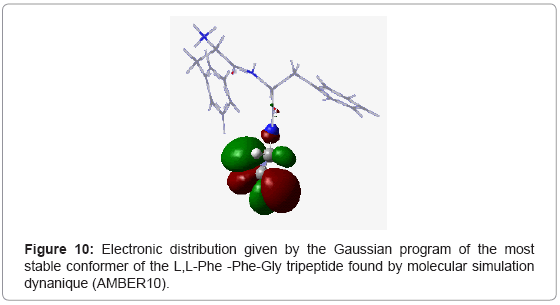
Figure 10: Electronic distribution given by the Gaussian program of the most stable conformer of the L,L-Phe -Phe-Gly tripeptide found by molecular simulation dynanique (AMBER10).
A complete understanding of tripeptides role in biological systems is largely dependent on the information available about the equilibrium mixture and the preferred conformation of the peptide molecules in solution. The conformational analysis offers a tool to determine all possible conformations which influence the dynamic behavior of tripeptides. This work represents a theoretical study of three oligopeptides: L,L-Gly-Phe-Phe, L,L-Phe-Gly-Phe, and L,L-Phe- Phe-Gly using molecular dynamic simulations in explicit water with the AMBER10 program package. RMSD measurement for the accuracy of peptides around their average conformations serves as an important indicator of many biological processes such as complex formations.
The presence of an aromatic ring in the C-terminal residue results into a more stable tripeptide compared to the tripeptide containing the same amino acids, but the residue phenyl is into N-terminal.