Journal of Developing Drugs
Open Access
ISSN: 2329-6631
ISSN: 2329-6631
Research Article - (2016) Volume 5, Issue 3
“Dracunculus medinensis” the Guinea Worm Disease (GWD) causing agent belong to the member of ‘Dracunculidae’ family. The infectious female nematode which is up to 800 mm (31 in) in length is the causative agent of the Guinea worm disease in the humans. Cyclops are the intermediate host of this infectious parasite. The healthy individual drinks or consume the contaminated water with Cyclops, which carries the infectious larvae of the Guinea Worm Disease. For the selection of the nonamers, the antigenic peptide the fragments of specific protein can be used, which can be further utilizing for the rational vaccine design and to increase the understanding the immune system response against the disease. The encouraging outcomes of the MHCII (Major Histocompatibility Complex II) analysis shows that the antigenic peptide of the Guinea Worm Disease are important determinant for the protection of the host from parasitic infection. In this study, the Position Specific Scoring Matrices (PSSM) and Support Vector Machine (SVM)-algorithms has been use for antigenic design and prediction of the binding affinity of the antigen having the 88 amino acids long residue, which shows 80 nonamers. The binding ability of the antigen to the major histocompatibility complex (MHC) class I and II molecules prediction will be helpful in the near future for specific targeted drug designing for the Guinea Worm Disease.
<Keywords: Antigen protein, Epitope, PSSM, SVM, MHC, Peptide, Vaccine
The long-established human infection which needs to be eradicated after the small pox is guinea worm disease. Guinea worm or Dracunculus medinensis considered as the one of most neglected tropical disease. Like other filarial nematode guinea worm completes its six developmental stages of life cycle with incubation period of approximately more than a year. The clinical importance makes this disease eradication as an urgent call [1]. The causative agent ‘Dracunculus’, is the only human infecting species. The Cyclops ingests the larvae of Dracunculus medinensis parasite, which are further ingested by the human from the contaminated stagnant water sources. The Cyclops digested by stomach digestive juices and led to release of the larvae. These larvae travel although and penetrate the digestive wall of the human and get entry into the abdominal cavity and retroperitoneal space. These larvae mature in adults and soon after the copulation the female mature and grows in size up to 60 cm to 3 m in size, whereas, the male dies. A year after the incubation period, the mature female worm come towards the skin subcutaneous tissue and start formation of a small round bulge on the skin, generally on the distal lower extremity and start secreting an irritating chemical. The symptoms arise as slight fever, local skin-redness, swelling and severe pruritus around the blister. Along with this the other symptoms include diarrhea, nausea, vomiting and dizziness. The blister burst within 1 to 3 days and female worms one or more slowly comes out from the wounds which causes an excessive burning sensation and pain [2]. Immersing or pouring water over the blister provide pain reliever. But this the moment that adult female is exposed to the external environment [3]. When infected individual emerges their limbs in the open source of water, the pathogenic parasite recognizes the difference of temperature and releases the milky white liquid in the water which contains millions of immature larvae, when larvae released in water are ingested by copepods where they mount twice and become infective larvae within two weeks [4]. The D. medinensis antigen peptides can be most suitable segment for the subunit vaccine development because of the fact that the immune response can be generated in large population with the single epitope. In the recent study we have used utrophin protein from Dracunculus medinensis to envision the antigenicity and solvent accessible regions that allows potential drug targets to identify active sites against versions reactions. Antigenicity prediction methods predicts those segments within utrophin that are antigenic and can elicited an antibody response. The N- and C- terminal region peptide prediction of protein is an important because these terminal regions of proteins are usually solvent accessible and unstructured, henceforth, antibodies against those regions are also likely to recognize the native protein that can help to design of synthetic peptide vaccine and immuno-diagnostic reagents [5-12].
Selection of desire data from database
The utrophin protein sequences from D. mednensis were retrieved from www.ncbi.nlm.nih.gov, UniProt databases for the further analysis [13-15] (Table 1).
| DVEVVKAQFKEHEQFMQSLTESQDSVGRVLHRGNVICQKLDDEQNMSLLSQLKLVNAKWERVREIAMNRQ NLLLEKLNSLQIQQLKKL |
Table 1: Utrophin, partial (Dracunculus medinensis) protein sequence retrieved from the NCBI database.
Computing the physical parameters of the protein
The retrieved sequence of the utrophin from Dracunculus medinensis were computed for its physical parameter with the help of the protpram [16] and recorded.
Recognition of protein antigenicity
Antigenecity prediction program were used to, predict the antigenicity of protein which predicts only those segments from protein that are capable to be antigenic by eliciting an antibody response. The methods used are Welling, Parker, Kolaskar and Tongaonkar antigenicity methods [17-25].
MHC class I binding peptide predictions
The neural networks trained on C terminals of known epitopes used for the MHC binding peptide prediction. The MHC class I binding peptide was predicted through RANKPEP [26] from the target protein sequences with the use of Position Specific Scoring Matrices (PSSMs) based approach (Table 2).
| Protein Name | Gene ID | No. of amino acids | Molecular Weight | Theoretical PI | Total number of atoms | Instability index | Aliphatic index | Grand average of hydropathicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|
| Utrophin, partial | GI:298919602 | 88 | 103300.9 | 8.11 | 1472 | 32.65 (This classifies the protein as stable) | 105.11 | -0.56 |
Table 2: Predicted physico-chemical properties of utrophin, partial Protein Sequence from (D. medinensis ).
MHC class II binding peptide predictions
The neural networks based approached is used to predict the MHC class II binding peptides from D. medinensis , which is trained well on c-terminal of known epitopes. We also predicted the peptide binders from the protein sequence by using RANKPEP. After the proteolytic cleavage the generated peptide fragments binds to the MHC molecule (Figure 1).

Figure 1: Hydrophobicity plot of antigen protein by Hphob/ Eisenberg et al. scale. At the Position: 45 the maximum Score: 0.026 (max) and at Position: 61 Score: -0.370 (min)
Cascade SVM based tappred method based antigenic peptide prediction
The high binding affinity peptides were predicted via Tappred methods which has potential to predict the highest affinity TAP binders on the basis of sequence and the properties of amino acids. Through this analysis we were able to predict the 26 high predicted affinity binder from the Dracunculus medinensis protein utrophin having the 88 amino acids, which shows 80 nonamers (Table 3).
| n | Start Position | Sequence | End Position |
|---|---|---|---|
| 1 | 22 | SQDSVGRVLHRGNVICQK | 39 |
| 2 | 46 | MSLLSQLKLVN | 56 |
| 3 | 70 | QNLLLEKLNSLQIQ | 83 |
Table 3: The antigenic determines of the protein sequence of utrophin from D. medinensis.
Prediction of the physico-chemical properties of the protein sequence
The targeted protein sequence of utrophin were retrieve from the NCBI database (Table 1) and analyzed for the physic-chemical properties. The physico-chemical properties like molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient [27-29], estimated half-life [30,31], instability index [32], aliphatic index [33] and grand average of hydropathicity (GRAVY) [34] were analyzed by the ProtParam [35]. Effect of temperature on protein solubility and denaturation. This physiochemical property prediction will helpful to understand the effect of pH on protein solubility and protein isoelectric point, interaction between protein and water molecules and hydrogen bonds, protein-ligand binding affinity, function of the protein. These property analysis plays an important role and taken into the consideration in the drug development or designing. The predicted Physio-chemical properties of the utrophin protein are shown in the Table 2.
Prediction of hydrophobicity
Eisenberg et al. method predicts antigenic determinants by searching protein sequences of utrophin from Dracunculus medinensis to find the area of greatest local hydrophilicity and the hydrophilic regions in the protein are located on the surface and are potentially antigenic. The point of highest local average hydrophilicity is located in or adjacent to an antigenic determinant. In this scale the amino acid value is starting from -3 (most hydrophobic) to 3 (most hydrophilic).
Welling antigenicity prediction method
Welling antigenicity method is based on the percentage of each amino acid present in known antigenic regions (utrophin from Dracunculus medinensis) compared to the percentage of the amino acids in the average composition of a protein. Previous strategies are based on the assumption that antigenic regions are primarily hydrophilic at the surface of the protein. This method is better than the Hopp-Woods scale of hydrophobicity which is also used to identify antigenic regions (Figure 2).

Figure 2: Hydrophobicity plot of antigen protein by Hphob/Welling et al. scale. The Min: -0.667, Max: Score: 0.636 (max) score at (position 63-65, 51-55) window-11.
Parker hydrophilicity prediction
Parker scale predicts antigenicity by identifying regions of greatest native hydrophilicity of utrophin from Dracunculus medinensis . It was derived from the Hopp-Woods scale however; these uses the HPLC retention times of model peptides to determine hydrophilicity. Parker hydrophilicity scale is sequence-based method that has been shown recently to perform prediction of linear epitopes of utrophin from Dracunculus medinensis (Figure 3).

Figure 3: Parker antigenicity plot of utrophin from Dracunculus medinensis hydrophilic scale based (threshold setting=1.396). Minimum: -2.111 (Position: 76) Maximum: 5.356 (Position: 24) the window size 7.
Kolaskar and Tongaonkar antigenicity
Kolaskar and Tongaonkar Antigenicity is a semi-empirical method for the prediction of antigenic regions including information of surface accessibility and flexibility. The method was able to predict antigenic determinants with an accuracy of 75% (Figure 4; Table 3).

Figure 4: Kolaskar and Tongaonkar Antigenicity Plot.
MHC class I and II binding peptide prediction
We found the binding of peptides to a number of different alleles using Position Specific Scoring Matrix. MHC molecules are cell surface proteins, which actively participate in host immune responses to almost all antigens. We have been able to predict MHC-I peptide binders of utrophin from D. medinensis . We found predicted MHC-I peptide binders of protein for Matrix: 8mer_H2_Db.p.mtx, Consensus: QNWNCCTI, Optimal Score: 52.494, Binding Threshold: 33.04; Matrix: 9mer_H2_Db.p.mtx, Consensus: FCIHNCDYM, Optimal Score: 50.365, Binding Threshold: 17.96; Matrix: 10mer_H2_Db.p.mtx, Consensus: SGYYNFFWCL, Optimal Score: 58.858, Binding Threshold: 41.32; Matrix: 11mer_H2_Db.p.mtx, Consensus: CGVYNFYYCCY, Optimal Score: 79.495, Binding Threshold: 56.96 (Table 4) and MHC-II peptide binders for Matrix: I_Ab.p.mtx, Consensus: YYAPWCNNA, Optimal Score: 35.632, Binding Threshold: 9.52; Matrix: I_Ad.p.mtx, Consensus: QMVHAAHAE, Optimal Score: 53.145, Binding Threshold: 7.10; Matrix: I_Ag7.p.mtx, Consensus: WYAHAFKYV, Optimal Score: 40.873, Binding Threshold: 7.54 for MHC II allele (Table 5) was tested and opted the result.
| MHC-I Allele | Rank | POS. | N | Sequence | C | MW (Da) | Score | % OPT. |
|---|---|---|---|---|---|---|---|---|
| 8mer_H2_Db | 1 | 33 | LHR | GNVICQKL | DDE | 856.04 | 10.238 | 19.50% |
| 8mer_H2_Db | 2 | 29 | VGR | VLHRGNVI | CQK | 889.06 | 7.466 | 14.22% |
| 8mer_H2_Db | 3 | 58 | VNA | KWERVREI | AMN | 1074.29 | 3.199 | 6.09% |
| 8mer_H2_Db | 4 | 23 | TES | QDSVGRVL | HRG | 854.96 | 3.11 | 5.92% |
| 8mer_H2_Db | 7 | 55 | LKL | VNAKWERV | REI | 960.13 | 0.848 | 1.62% |
| 9mer_H2_Db | 1 | 41 | QKL | DDEQNMSLL | SQL | 1046.12 | 10.305 | 20.46% |
| 9mer_H2_Db | 2 | 32 | VLH | RGNVICQKL | DDE | 1012.23 | 8.094 | 16.07% |
| 9mer_H2_Db | 3 | 28 | SVG | RVLHRGNVI | CQK | 1045.25 | 7.847 | 15.58% |
| 9mer_H2_Db | 4 | 64 | RVR | EIAMNRQNL | LLE | 1070.23 | 6.858 | 13.62% |
| 9mer_H2_Db | 5 | 65 | VRE | IAMNRQNLL | LEK | 1054.27 | 3.794 | 7.53% |
| 10mer_H2_Db | 1 | 52 | LSQ | LKLVNAKWER | VRE | 1215.49 | 8.991 | 15.28% |
| 10mer_H2_Db | 2 | 68 | IAM | NRQNLLLEKL | NSL | 1222.45 | 5.042 | 8.57% |
| 10mer_H2_Db | 3 | 63 | ERV | REIAMNRQNL | LLE | 1226.42 | 1.07 | 1.82% |
| 10mer_H2_Db | 4 | 10 | AQF | KEHEQFMQSL | TES | 1258.42 | 0.713 | 1.21% |
| 10mer_H2_Db | 5 | 6 | EVV | KAQFKEHEQF | MQS | 1273.42 | 0.692 | 1.18% |
Table 4: Promiscuous MHC ligands, having C-terminal ends are proteosomal cleavage sites of D. medinensis (All rows highlighted in red represent predicted binders and A peptide highlighted in violet has a C-teminus predicted by the cleavage model used).
| MHC-II Allele | Rank | POS. | N | Sequence | C | MW (Da) | Score | % OPT. |
|---|---|---|---|---|---|---|---|---|
| MHC-II I_Ad | 1 | 3 | DV | EVVKAQFKE | HEQ | 1059.23 | 15.79 | 29.71% |
| MHC-II I_Ad | 2 | 15 | HEQ | FMQSLTESQ | DSV | 1052.17 | 12.824 | 24.13% |
| MHC-II I_Ad | 3 | 28 | SVG | RVLHRGNVI | CQK | 1045.25 | 11.195 | 21.07% |
| MHC-II I_Ad | 4 | 52 | LSQ | LKLVNAKWE | RVR | 1059.3 | 5.345 | 10.06% |
| MHC-II I_Ad | 5 | 6 | EVV | KAQFKEHEQ | FMQ | 1126.24 | 3.863 | 7.27% |
| MHC-II _Ag7 | 1 | 47 | QNM | SLLSQLKLV | NAK | 982.23 | 13.281 | 32.49% |
| MHC-II _Ag7 | 2 | 67 | EIA | MNRQNLLLE | KLN | 1112.31 | 5.691 | 13.92% |
| MHC-II _Ag7 | 3 | 25 | SQD | SVGRVLHRG | NVI | 962.12 | 5.593 | 13.68% |
| MHC-II _Ag7 | 4 | 61 | KWE | RVREIAMNR | QNL | 1126.35 | 5.119 | 12.52% |
| MHC-II _Ag7 | 5 | 80 | LNS | LQIQQLKKL | 1093.37 | 4.883 | 11.95% | |
| MHC-II _Ak | 1 | 42 | KLD | DEQNMSLLS | QLK | 1018.11 | 10.177 | 25.51% |
| MHC-II _Ak | 2 | 1 | DVEVVKAQF | KEH | 1016.16 | 9.884 | 24.77% | |
| MHC-II _Ak | 3 | 31 | RVL | HRGNVICQK | LDD | 1036.21 | 9.375 | 23.50% |
| MHC-II _Ak | 4 | 68 | IAM | NRQNLLLEK | LNS | 1109.29 | 9.144 | 22.92% |
| MHC-II _Ak | 5 | 12 | FKE | HEQFMQSLT | ESQ | 1102.23 | 8.899 | 22.30% |
Table 5: Prediction of MHC-II ligands all rows highlighted in red represent predicted binders to the MHC-II Allele i.e., MHC-II I_Ab, MHC-II I_Ad, MHC-II I_Ag7, I_Ak.
Antigenic peptides prediction by cascade SVM based TAPPred method
The cascade SVM based TAPPred method were also used and found 105 High affinity TAP Transporter (Transporter peptide regions) (Tables 6 and 7) peptide regions which represents predicted TAP binder’s residues which occur at N and C termini from D. medinensis antigenic utrophin. TAP is an important transporter that transports antigenic peptides from cytosol to ER. The efficiency of TAP-mediated translocation of antigenic peptides is directly proportional to its TAP binding affinity. Thus, by understanding the nature of peptides, that bind to TAP with high affinity, is significant steps in endogenous antigen processing. The correlation coefficient of 0.88 was received by using jackknife validation test. T cell immune responses are derived by antigenic epitopes; hence, their identification is important for design synthetic peptide vaccine.
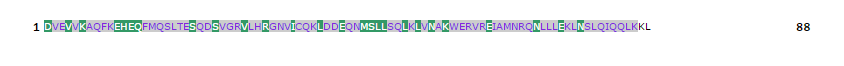
Table 6: Cascade SVM based High affinity TAP Binders display format results, where the results are shown by colouring the residues. The green colour background of the residues demarcates the N terminal of residue. The rest residues of peptide are shown with violet-blue background.
| Peptide Rank | Start Position | Sequence | Score | Predicted Affinity |
|---|---|---|---|---|
| 1 | 25 | SVGRVLHRG | 8.647 | High |
| 2 | 52 | LKLVNAKWE | 8.628 | High |
| 3 | 11 | EHEQFMQSL | 8.624 | High |
| 4 | 12 | HEQFMQSLT | 8.603 | High |
| 5 | 32 | RGNVICQKL | 8.552 | High |
| 6 | 75 | EKLNSLQIQ | 8.546 | High |
| 7 | 6 | KAQFKEHEQ | 8.523 | High |
| 8 | 29 | VLHRGNVIC | 8.468 | High |
| 9 | 58 | KWERVREIA | 8.457 | High |
| 10 | 14 | QFMQSLTES | 8.422 | High |
Table 7: Cascade SVM based High affinity TAP Binders results in the tabular format.
The utrophin antigenic proteins from Dracunculus medinensis are involved in various antigenic components too direct and empower the immune system to protect host from the infection. The MHC class I and class II specifically binds to their respective epitopes. In host immune responses these surface proteins (MHC) actively participate against almost all the antigen and provide protection to the host from infections. The know-how concept of the immune response against an antigen conclude the concept, that the whole protein is not required to elicited the immune responses, but only the modest fragment of antigen is also capable to stimulate the immune responses against the pathogen or whole antigen. This phenomenon shows that by increasing in the affinity of MHC binding peptides may result in enhanced immunogenicity of utrophin from the D. medinensis , hence may anticipated in silico drug designing and might be helpful in the development of the more advanced highly predictive computational tools for the better identification of the T cell epitopes. Last but not the least, the accurate prediction remains important for the future synthetic peptide vaccine designing.