Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2013) Volume 0, Issue 0
Herpes simplex virus causes multiple infections through genetic and proteomic material. In current scenario broad spectrum antibiotic used to control and reduce the infection. This paper is the focus on the development of structure-based drug design for HHV infection that involves choosing the target proteins, visualizing the target structure, identifying the binding site, docking the ligands, and evaluating those using computational techniques. Homology modeling performs for Protein Database (PDb) structure of glycoprotein and DNA polymerase of HHV-I and II. On the basis of their known antiviral activity, twenty one natural molecules select for proposed drug. Finally, 15 molecules pass the Lipinski rule of five for ligand selection and best of that based on Ligand Efficiency, Binding Affinity and Inhibitory Constant propose for drug target.
<Keywords: HHV, Homology modeling, Docking, Lipinski rule ligand efficiency, Binding affinity, Inhibitory constant
Three dimensional structures of bimolecular targets have increased dramatically because the structural and electronic properties about ligands have also increased. This has inspired the rapid development of the structure-based drug design. Structure-based drug design is the basis on statistics of the 3-D structure of the biological target. Currently structure-based drug design can be divided into two categories. The first class is ligand-based drug designing, in this case, a possible large number of ligand molecules are select to get the best for fit in the proteins pocket of the receptor. The main benefit of database searching is that it saves computational time to acquire new lead compounds. The second group of structure-based drug design methods is known as receptor-based design. In this case, ligand molecules are select within the constraints of the binding pocket. The key role of such a system is that novel structures can formulate, that not contain in any database.
The Structure based drug design having five steps. In a first step, identification of drug target or a receptor is a macromolecule that is critical for a disease causing through genetic or proteomic material. Second step involves accurate structural information of biomolecules to understand the function and activity of drug target. X-ray Crystallography (XRC), Nuclear Magnetic Spectroscopy (NMR), and homology modeling are the common techniques use for structural information of proteins. Next step is the identification of the binding site at which ligand can attach and inhibit the disease causing proteins. In second last step, computational studies use to evaluate the inhibitor binding complex. Most work falls into the two categories namely Docking and Scoring. Final step applies for evaluation of potential lead compound before proceeding to the preclinical trials.
The drug target in structure-based drug design is mostly proteins. The proteins belong to families of glycoprotein and DNA polymerase. The most popular method for identifying drug targets is by the study of the disease under observation and identification of genomics and proteomics associated with the disease. The vast amount of biological data is a treasure to identification and characterization of virulent factors in pathogens, and to identify novel putative targets for therapeutic intervention [1]. The strategies for drug design and development are progressively shifting from the one era to another in the presence of biological information [2]. Worldwide rates of HSV infection is between 65–90 percent. HSV1 is more prevalence than HSV2 with rates of both increasing as people age. An estimated 536 million people worldwide were infected with HSV-2 in 2003, with the highest rates in sub-Saharan Africa and the lowest rates in Western Europe [3]. Analgesics (ibuprofen and acetaminophen) can reduce pain and fever caused by viral infection of HSV. Topical anesthetic treatments such as prilocaine, benzocaine or tetracaine can also relieve itching and pain [4-6]. There are several antiviral that are effective for treating herpes including acyclovir and valacyclovir. Aciclovir and Valacyclovir are available as a generic medicine [7,8]. Evidence supports the use of aciclovir and valaciclovir in the treatment of herpes labialis [9] as well as herpes infections in people with cancer [10]. However, the previous result shows that use of acyclovir for herpetic gingivostomatitis is less strong [11]. Overall there is no method to eradicate herpes virus from the body, but antiviral medications can reduce the frequency, duration, and severity of outbreaks.
Homology modeling of proteins is necessary for ligand design and literature reveal that identification and visualization of protein cavities is the starting point for many Structure-Based Drug Design (SBDD) applications [12]. The size and shape of protein cavities dictates the three-dimensional geometry of ligands that can strongly bind and its minimal requirement for drug activity [13]. The homology modeling method takes its inspiration from calculations that require to construct a three-dimensional structure from data sequence and using spatial restraint-based modeling software Modeller [14]. DOPE is using as reference state that corresponds to non interacting atoms in a homogeneous sphere with the radius dependent on a sample original structure. The Ramachandran plot is a fundamental tool in the analysis of protein structures and the interactions of the Glycine and pre-proline. Both these methods are applicable for optimization of model [15,16]. Molinspiration molecule viewer allows visualization of collection of molecules encoded as SMILES or SD file of antiviral compound and Lipinski’s rule of five for prediction of oral active drug [17]. Autodock is an automated docking tool that was designed to predict how small molecules bind to receptor of known 3D structure and it also optionally enables to model Binding parameters of ligand with a number of distinct conformational clusters and to find all possible minimum binding energy [18].
As discuss in the end of the first section in the literature review only antiviral compound decrease the infection in homosapiens. Viral capsid proteins of HHV cause severe infection to a human being through DNA maturation and glycoprotein. The objective of this paper is to design drug using comparative modeling for oral indigestion using computational and bioinformatics tool. Another core area of the paper is to propose antiviral natural compounds.
Structure-based drug design has tremendous potential and an alternative to conventional screening techniques. This drug design method involves bioinformatics, proteomics, biochemistry, and computer modeling of 3-dimensional protein structures. Whole proteomic sequences (DNA polymerase and Envelope glycoprotein B) were downloaded for Human herpes virus 1 and 2 from NCBI. The GI numbers of four possible proteins are 386089622, 384597774, 386089624 and 360039889, for comparative modeling using MODELLER 9.9. The best model chooses on the basis of DOPE Score and RC Plot analysis. Pymol help to eliminate unwanted part of primary chain and q server request for pocket identification. Propose antiviral compounds properties see using Molinspiration and Lipinski rule of five. Optimized ligands are uses to get appropriate interactions with the target protein. The docking of the ligands with the target protein performs using the autodock.
Homologous modeling
The three-dimensional structure of (pdb) of DNA polymerase and glycoprotein model develop by considering the suitable well studied template proteins structure identify by similarity search with the blast tool against the protein databank. These selected proteins align with matrix Blosum 60, gap existence 7.0 and gap extension 0.4 against the database of related proteins. We select ten possible models for glycoprotein using modeller 9.9 as shown in Table 1. Ramachandran plot analysis for same 20 glycoproteins of HHV-1 and HHV-2 is representing in Table 2.
| Glycoprotein of HHV1 Filename | molpdf | DOPE score | Glycoprotein of HHV2 Filename | molpdf | DOPE score |
|---|---|---|---|---|---|
| seq.B99990001.pdb | 7299.93164 | -63119.12500 | seq.B99990001.pdb | 8086.02441 | -66823.78125 |
| seq.B99990002.pdb | 7456.70459 | -63039.52734 | seq.B99990002.pdb | 8007.50049 | -66583.03906 |
| seq.B99990003.pdb | 6910.33008 | -63272.28516 | seq.B99990003.pdb | 8032.51416 | -66737.57031 |
| seq.B99990004.pdb | 6795.05029 | -63459.29297 | seq.B99990004.pdb | 7658.65381 | -65636.45313 |
| seq.B99990005.pdb | 6395.35742 | -65302.81250 | seq.B99990005.pdb | 7764.36523 | -66265.73438 |
| seq.B99990006.pdb | 7132.14258 | -63455.75391 | seq.B99990006.pdb | 7825.49219 | -67258.58594 |
| seq.B99990007.pdb | 7092.15869 | -63402.32813 | seq.B99990007.pdb | 10209.6455 | -64925.18750 |
| seq.B99990008.pdb | 6592.68701 | -63724.46094 | seq.B99990008.pdb | 8018.68408 | -66633.06250 |
| seq.B99990009.pdb | 6380.85742 | -63971.69531 | seq.B99990009.pdb | 7660.81201 | -66978.28906 |
| seq.B99990010.pdb | 6861.02441 | -64183.12500 | seq.B99990010.pdb | 7925.67627 | -66644.86719 |
Table 1: Output (molpdf and dope score) of Protein structure modeling by satisfaction of spatial restraints of glycoprotein’s of HHV1 and HHV-2.
| Sequence No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| HHV-1 | 93.1 | 92.9 | 94.7 | 94.6 | 94.0 | 93.5 | 93.6 | 93.5 | 92.5 | 94.9 |
| HHV-2 | 94.3 | 94.9 | 95.1 | 94.2 | 95.1 | 93.7 | 92.5 | 95.7 | 96.3 | 95.1 |
Table 2: RC Analysis of Glycoprotein of HHV-1 and HHV2 for percentage of residues in favored regions.
The best model selected with the lowest value of the MODELLER objective function, the DOPE assessment score, with the highest percentage residue core region from Ramachandran plot. On the basis of results in Table 2, seq.B99990010.pdb and seq.B99990009.pdb of glycoprotein put on rank 1 in respective strains. Similarly 94.4% of favored residues are present in DNA polymerase of HHV-2 species. Again the MODELLER is to use the variability in the loop region from the three templates to generate a more accurate conformation of the loop.
However, the conformation of a loop in the region around the residue at the C-terminal end of the sequence has higher DOPE score than for the model based on a single template. The RC analysis for residues of favored region of DNA polymerase is 91.1 and 92.9% respectively for HHV-1 and II. Glycoprotein has favour residues 93.5 and 93.7 % of propose pdb structure of target of HHV-1 and II strain shown in Figure 1.
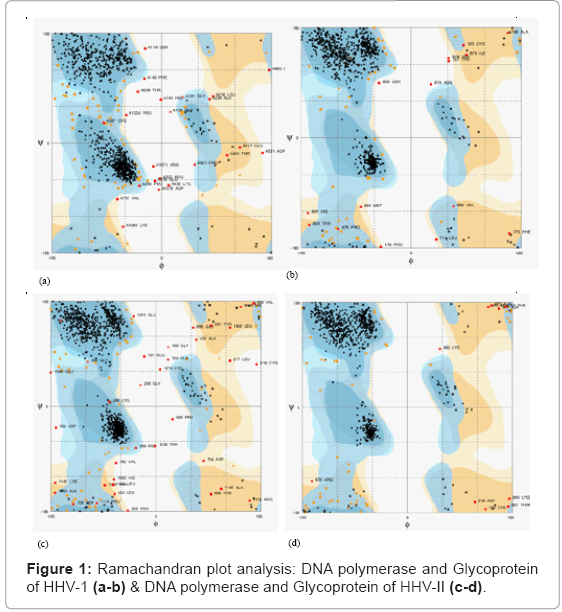
Figure 1: Ramachandran plot analysis: DNA polymerase and Glycoprotein of HHV-1 (a-b) & DNA polymerase and Glycoprotein of HHV-II (c-d).
The modeled protein is validated with Pymol, and total energy values of the predicted 3-D model were calculated as 91.1% of Ramachandran plot for DNA polymerase of HHV-1. Active sites of the target protein were predicted by q server active site prediction tools and propose antiviral compound listed in Table 3 and ligand properties are collected from NCBI Pubchem Compound database (Figure 2).
| Molecule Number | CID | Polar surface area | Number of atoms | Rotatable bonds | Volume (Angstroms) |
|---|---|---|---|---|---|
| 1. | 65036 | 17.071 | 9.0 | 5 | 145.506 |
| 2. | 87310 | 80.393 | 11.0 | 5 | 154.764 |
| 3. | 5386591 | 17.071 | 13.0 | 8 | 207.853 |
| 4. | 108058 | 118.36 | 39.0 | 8 | 488.958 |
| 5. | 370 | 97.983 | 12.0 | 1 | 135.098 |
| 6. | 65084 | 130.602 | 22.0 | 1 | 252.159 |
| 7. | 5281764 | 208.12 | 34.0 | 11 | 385.951 |
| 8. | 53398694 | 161.587 | 22.0 | 7 | 251.14 |
| 9. | 442793 | 66.761 | 21.0 | 10 | 295.61 |
| 10. | 5281794 | 46.533 | 20.0 | 9 | 281.379 |
| 11. | 1309 | 66.479 | 28.0 | 3 | 333.361 |
| 12. | 2353 | 40.821 | 25.0 | 2 | 296.302 |
| 13. | 99474 | 38.696 | 30.0 | 0 | 419.383 |
| 14. | 305 | 20.228 | 7.0 | 2 | 120.158 |
| 15. | 5280951 | 34.153 | 16.0 | 1 | 201.15 |
| 16. | 5280953 | 37.917 | 16.0 | 1 | 194.963 |
| 17. | 92097 | 20.228 | 31.0 | 0 | 460.699 |
| 18. | 5281792 | 144.516 | 26.0 | 7 | 303.539 |
| 19. | 10364 | 20.228 | 11.0 | 1 | 158.572 |
| 20. | 5281515 | 0.0 | 15.0 | 0 | 229.95 |
| 21. | 12575 | 66.761 | 12.0 | 2 | 144.608 |
Table 3: Molecular properties of 21 natural compounds calculated by Molinspiration with Chemical identifier for molecular structures in Pubchem.
Figure 2: Modeled protein ribbon structure of DNA polymerase and surface structure shown in a & b respectively.
An antiviral compound
We select the 21 compounds for anti viral activity from natural resources such as Garlic (Allium sativum), Azadirachta indica, Echinacea, Ginger (Zingiber officinale), Goldenseal, Methika (Fenugreek), Peganum harmala, Clitorea ternatea, Ocimum sanctum and Scrophularia frutescense. Molinspiration tool use to evaluate the physical and chemical properties of propose anti viral compounds listed in Table 3 with CID number as in Pubchem. Molinspiration calculate the molecular Polar Surface Area (PSA), molecular volume, number of rotatable bonds and number of atoms as shown in Table 3. Similarly Lipinski rule of five to use to check the violation the rule of the drug is representing in Table 4. Polar surface area of molecules related to transport of drug, in our result it has been observing that molecules number 4, 6, 7, 8 and 10 having polar surface value greater than 100. Even the molecule number eight has polar surface area 208.12, observed highest in the 21 natural compounds. Molecular volume signifies the intestinal absorption or blood-brain barrier penetration of drug. The molecular volume above than 300 units found in the compounds 4, 7, 11, 13 and 17 and highest value among five is for compound number four. Rotatable bond covers the molecular flexibility and oral bioavailability of drugs. Only two compounds (7 and 9) have significant value of rotatable bond that is exceeding than 10. Optimization of leads compound based on the Lipinski rule of five, in these poorly soluble compounds as well as insoluble and, nonpermeable compounds would have been filter out at earlier stages. Out of 21 viable natural compounds, five compounds violate the Lipinski rule of five. These compounds are places at position number 4, 6, 7, 13, 17 and 20 in Table 4. After rejection of these six compounds only 15 compounds may use for optimization of lead compound.
| Molecule Number | CID | Molecular Weight |
Log P | H-donor | H-acceptor | Violations |
|---|---|---|---|---|---|---|
| 1. | 65036 | 162.279 | 2.064 | 1 | 0 | 0 |
| 2. | 87310 | 177.225 | -3.393 | 4 | 3 | 0 |
| 3. | 5386591 | 234.411 | 1.802 | 1 | 0 | 0 |
| 4. | 108058 | 540.609 | 3.552 | 9 | 0 | 1 |
| 5. | 370 | 170.12 | 0.589 | 5 | 4 | 0 |
| 6. | 65084 | 306.27 | 1.077 | 7 | 6 | 1 |
| 7. | 5281764 | 474.374 | 1.269 | 12 | 6 | 2 |
| 8. | 53398694 | 312.23 | -0.608 | 9 | 5 | 0 |
| 9. | 442793 | 294.391 | 3.217 | 4 | 2 | 0 |
| 10. | 5281794 | 276.376 | 4.348 | 3 | 1 | 0 |
| 11. | 1309 | 383.4 | 2.831 | 7 | 0 | 0 |
| 12. | 2353 | 336.367 | 0.196 | 5 | 0 | 0 |
| 13. | 99474 | 414.63 | 5.932 | 3 | 1 | 1 |
| 14. | 305 | 104.173 | -4.236 | 2 | 1 | 0 |
| 15. | 5280951 | 214.268 | 2.505 | 3 | 1 | 0 |
| 16. | 5280953 | 212.252 | 2.626 | 3 | 1 | 0 |
| 17. | 92097 | 426.729 | 8.023 | 1 | 1 | 1 |
| 18. | 5281792 | 360.318 | 1.626 | 8 | 5 | 0 |
| 19. | 10364 | 150.221 | 3.815 | 1 | 1 | 0 |
| 20. | 5281515 | 204.357 | 5.174 | 0 | 0 | 1 |
| 21. | 12575 | 168.148 | 1.187 | 4 | 2 | 0 |
Table 4: Lipinski Rule of 5 of 21 natural compounds calculated by Molinspiration with Chemical identifier for molecular structures in Pubchem.
Molecular docking
Molecular docking use the computational simulation which predicts the preferred orientation of ligand to a receptor when interact each other to form a higher stability complex. In this paper, rigidflexible docking is performing using Autodock 4.2 tool, to predict the binding affinity, ligand efficiency and inhibitory constant. It gives 10 different conformations of the ligand molecule at the pocket of the protein molecule. The results of Autodock for DNA polymerase (HHV-I and HHV-II) are representing in tables 5 and 6. Ligand Efficiency (LE) measures the ratio of Gibbs free energy to the number of nonhydrogen atoms of the compound and represent in the negative magnitude. The molecule number 19 having maximum ligand efficiency -0.48 and corresponding lowest binding affinity (BI) -4.46 in HHV-I. Similar results are observed for molecule number 10 that shows the LE value -0.47 and BI equal to -8.31 as compare to molecule number 19. The inhibitory constant in both case is found 637.57 uM and 817.12 mM respectively for HHV-I and II.
Ligand Efficiency (LI) of all the molecules lies in the range of -0.01 to -0.31 for HHV-I species. The extreme value of LI observes for molecules place at number 2. Complimentary to LI the BI value also found in uppermost level -5.17. HHV-2 species have binding affinity lies in the array of -0.6 to -6.31. Molecule number 14 shows the maximum values of LI efficiency -0.41. Moreover, the inhibitory constants for HHV-I and HHV-II are 162.74 uM & 23.85 uM respectively. All the results of docking at RMSD value of zero angstrom.
Homology models used to assess protein structure prediction using DOPE potential, which accounts the finite and spherical shape of the native structures. SeqB99990010.pdb and seq-B99990009. pdb of glycoprotein, having dope energy respectively -64183.12500 and -66978.28906. The percentage of favored residues according to Ramachandran plot is also highest in both pdb structures. Their values for RC analysis are 94.9 and 96.3 support on the pdb structure for further analysis. DNA polymerase of HHV-I structure exists in the literature so homology modeling for these proteins is not required, but corresponding to lowest dope value of HHV-II the RC value is 94.7%. Water molecules and other secondary chains of that pdb structure remove to focus on primary chain of proteins. The RC percentage of favored residues in the range of 91.1 and 92.9 (DNA polymerase) and Glycoprotein have favor residues percentage between 93.5- 93.7%. Such higher values support the quality of a structure modeling.
Molecule polar surface area of propose drug lies in the region of 0 to 208.12, which relate to human intestinal absorption capacity. Molecules no 7 has highest the PSA value indicates that probability to absorb the drug is top. This property arises because of the sum of surface of polar atoms is also highest in compound 7. The number of atoms varies from seven to thirty four in all the natural compounds. Other molecular properties of compounds are molecular volume and rotatable bonds present in the drug. Molecular volume also main concern with transport characteristics of molecules, in our simulated results the values of volume vary from 144.608 to 488.958. In this case, the higher molecular volume is found for molecule number 4. All the molecules show the volume in viable range due to flexibility of rotatable atoms. Other significant advantage of rotatable atoms to descriptor of oral bioavailability of drugs, so from Table 3 good indication is achieving for structure drug design. Lipinski rule of five applies to check the validity of natural compound. According to this rule (log ≤ 5, MW ≤ 500, number of hydrogen bond acceptors ≤ 10, and number of hydrogen bond donors ≤ 5) only six molecules enlist in Table 4 are removing from the docking process. It means bioavailability of 15 molecules for drug design purpose.
Docking generally relies on ligand efficiency, binding affinity and inhibitory constant. Ligand efficiency and binding affinity are correlated terms because it solely depends upon free energy. LI increases with minimization of Gibbs free energy. On the other side binding, affinity maximizes with lowering of energy. Table 5 indicates that the molecules 19 (HHV-1) and 10 (HHV-2) have ligand efficiency around ¦0.50¦ which is most suitable for selection of lead compound. The results for the binding affinity for same compounds are at lowest, also support for further analysis. The inhibitory constant in both cases are 637.57 and 817.12 uM respectively for HHV-I and II. The results of docking test for glycoproteins are not too much different from Table 3. LI and BI values of HHV-I & II strains are ¦0.31¦, ¦0.41¦, -5.17 and -6.31 respectively. The molecule number 2 (HHV-I) & 14 (HHV-II) have inhibitory constant are 162.77 and 23.85 uM.
| HHV-1 | HHV-2 | ||||||
|---|---|---|---|---|---|---|---|
| Molecule No | Ligand Efficiency | Binding Affinity (kcal/mol) | Inhibitory Constant mM | Molecule No | Ligand Efficiency | Binding Affinity (kcal/mol) | Inhibitory Constant mM |
| 1. | -0.17 | -1.54 | 74.43 | 1. | -0.22 | -1.96 | 36.39 |
| 2. | -0.15 | -3.76 | 1.26 | 2. | -0.27 | -6.68 | 12.64 uM |
| 3. | -0.27 | -2.47 | 15.39 | 3. | -0.31 | -2.76 | 9.54 |
| 5. | -0.09 | -2.06 | 30.83 | 5. | -0.30 | -6.56 | 15.56 uM |
| 8. | -0.23 | -2.55 | 13.46 | 8. | -0.40 | -4.35 | 643.88 uM |
| 9. | -0.25 | -3.95 | 1.28 | 9. | -0.35 | -5.54 | 87.02 uM |
| 10. | -0.12 | -1.05 | 93.23 | 10.** | -0.47 | -8.31 | 817.12 uM |
| 11. | -0.20 | -2.39 | 17.79 | 11. | -0.29 | -3.48 | 2.79 |
| 12. | -0.07 | -0.97 | 114.83 | 12. | -0.17 | -3.82 | 1.54 |
| 14. | -0.13 | -0.56 | 99.78 | 14. | -0.15 | -3.13 | 6.43 |
| 15. | -0.28 | -4.43 | 564.79 uM | 15. | -0.35 | -5.54 | 87.34 uM |
| 16. | -0.04 | -1.17 | 138.41 | 16. | -0.24 | -6.72 | 11.79 uM |
| 18. | -0.04 | -0.82 | 249.23 | 18. | -0.24 | -4.78 | 315.78 uM |
| 19.* | -0.48 | -4.46 | 637.57 uM | 19. | -0.43 | -6.42 | 19.83 uM |
| 21. | -0.10 | -0.71 | 143.65 | 21. | -0.19 | -7.6 | 2.7 uM |
Table 5: Docking results of screened Natural Antiviral compounds with HHV-1 and HHV-2 of DNA polymerase.
| HHV-1 | HHV-2 | ||||||
|---|---|---|---|---|---|---|---|
| Molecule No | Ligand Efficiency | Binding Affinity (kcal/mol) | Inhibitory Constant mM | Molecule No | Ligand Efficiency | Binding Affinity (kcal/mol) | Inhibitory Constant mM |
| 1. | -0.12 | -1.12 | 151.26 | 1. | -0.09 | -1.08 | 357.89 |
| 2.* | -0.31 | -5.17 | 162.74 uM | 2. | -0.12 | -0.6 | 283.12 |
| 3. | -0.14 | -1.27 | 117.75 | 3. | -0.25 | -2.23 | 23.11 |
| 5. | -0.09 | -2.03 | 32.6 | 5. | -0.19 | -4.16 | 891.05 uM |
| 8. | -0.24 | -2.68 | 10.94 | 8. | -0.34 | -3.73 | 1.85 |
| 9. | -0.11 | -1.02 | 179.96 | 9. | -0.29 | -4.68 | 374.07 µM |
| 10. | -0.13 | -4.15 | 911.93 uM | 10. | -0.18 | -5.45 | 100.59 µM |
| 11. | -0.19 | -2.25 | 22.31 | 11. | -0.22 | -2.69 | 10.68 |
| 12. | -0.07 | -1.6 | 67.57 | 12. | -0.11 | -2.37 | 18.21 |
| 14. | -0.26 | -3.67 | 1.53 | 14.** | -0.41 | -6.31 | 23.85 µM |
| 15. | -0.17 | -2.79 | 8.97 | 15. | -0.31 | -4.9 | 255.87 µM |
| 16. | -0.15 | -4.17 | 874.94 uM | 16. | -0.15 | -4.27 | 740.21 µM |
| 18. | -0.08 | -1.53 | 76.18 | 18. | -0.18 | -3.67 | 2.03 |
| 19. | -0.26 | -3.88 | 1.42 | 19. | -0.34 | -5.17 | 161.72 µM |
| 21. | -0.01 | -0.36 | 545.97 | 21. | -0.11 | -4.27 | 743.19 µM |
Table 6: Docking results of screened Natural Antiviral compounds with HHV-1 and HHV-2 of Glycoprotein.
Drug design from sequences and computer-aided techniques (modeller, Autodock and Molinspiration) has become a vital component of bioinformatics to identify probable anti-viral compounds. These four natural compounds selected as ligands are able to control the infection in the human being through glycoprotein and DNA polymerase. It was found that ligand having CID number 10364, 5281794, 87310 and 305 were good inhibitors based on docked results. Lipinski’s rules also support four ligands for oral bioavailability. The real potential of the ligands and proof in this paper analysis need to be tested experimentally.