Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2015) Volume 4, Issue 2
Aim: The aim of the current study was to assess the possible protective effects of advanced glycation end product (AGE) inhibitors against liver fibrosis and the possible underlying mechanisms.
Material and Methods: The present study was conducted on 48 male Wistar albino rats that were grouped into six groups of 8 rats each. Groups I-III; normal groups that were injected intraperitoneally (I.P.) with normal physiological saline, and either received no treatment (Group I), or received aminoguanidine (group II) or lisinopril (group III) daily for 4 weeks by I.P. injection. Group IV included untreated liver fibrosis group in which liver fibrosis was induced by thioacetamide. Groups V and VI included treated liver fibrosis group in which aminoguandine and lisinopril were injected I.P., daily for 4 weeks, concomitantly with thioacetamide. At the end of the treatment period (4 weeks), serum was collected to measure aminotransferases (AST and ALT) activities. Hepatic levels of AGEs, transforming growth factor-B1 (TGF-β1), tissue inhibitor of matrix metalloproteinase (TIMP), malondialdehyde (MDA), reduced glutathione (GSH) and hydroxyproline levels were assessed. Histopathological examination of liver was also carried out.
Results: TAA administration resulted in hepatic fibrosis evidenced histologically and by a significant increase in hepatic hydroxyproline level. TAA also resulted in a significant increase in serum AST, ALT activities as well as hepatic AGEs, TGF-β1, MDA and TIMP concentrations, together with a significant decrease in hepatic GSH. Administration of either aminoguanidine or lisinopril resulted in significant amelioration of above mentioned parameters. Conclusion: Targeting AGEs could represent a therapeutic option for patients at risk for developing liver fibrosis.
Keywords: Advanced glycation end products; Lisinopril; Aminoguanidine; Thioacetamide; Fibrosis
Liver cirrhosis is a serious irreversible disease and is the tenth leading cause of death in developed countries. If treated properly at fibrosis stage, cirrhosis could be prevented. Several experimental ameliorative strategies have been tested. However, there is still a remarkable lack of definitive evidence supporting specific therapies in these settings that can either cure or at least halt the progress of liver injury to liver fibrosis. Therefore, better strategies and /or drugs are desperately needed that would reduce the risk of liver fibrosis.
Although the pathophysiology of liver cirrhosis is not completely understood, some key events leading to tissue injury and thereby to liver fibrosis have been identified [1]. Much attention has been paid in the recent years to the nonenzymatic glycation and advanced glycation end products (AGEs) hypothesis. This hypothesis has been issued based on the fact that diabetes is featured by hyperglycemia, which facilitates the formation of AGEs and diabetes is commonly accompanied by nonalcoholic steatohepatitis, which could cause hepatic fibrosis. Indeed, Elevated levels of serum AGEs were observed in patients with nonalcoholic steatohepatitis [2].
Non-enzymatic glycation involves the reaction of the carbonyl group of sugar aldehydes with the N-terminus of free amino groups of proteins, resulting in the formation of Schiff ’s base that is subsequently rearranged to amadori products in a reaction called Maillard reaction. These amadori products or glycated proteins may then react with other proteins, lipids or DNA resulting in irreversible crosslinking and the formation of AGEs (post-amadori or glycotoxins) [3]. Although AGEs formation happens as a result of normal aging, it occurs at an accelerated rate in diseases as diabetes mellitus [4], renal failure [5], diabetic nephropathy [6], inflammatory conditions [7] and liver cirrhosis [8]. Recently, AGEs have been reported to significantly increase in hepatic fibrosis and to play a critical role in stimulating extracellular matrix (ECM) synthesis [9].
Several possible mechanisms could account for the ability of AGEs to stimulate liver fibrosis. AGEs can increase oxidative stress; induce production of pro-inflammatory cytokines as tumor necrosis factor-α (TNF-α), and transforming growth factor-β (TGF-β) [3]. Effects of AGEs are mediated by their receptor system, which could be generally divided into two categories. Receptor for AGEs (RAGE) facilitates oxidative stress (OS), cell growth and inflammation [10] and AGE receptors (AGE-Rs), eg. AGE-R1 (also called OST-48), is responsible for detoxification and clearance of AGEs [11].
Inhibition of AGEs formation, blockade of AGEs-RAGE interaction, suppression of RAGE expression, interruption of its signaling and induction of AGE-R1 expression are, thus, novel therapeutic strategies for targeting AGE-mediated diseases [12,13].
AGE inhibitors, as aminoguanidine, carnosine, acarbose and pyridoxamine, vary widely in structure, but they have common mechanism of action, which is the trapping of reactive carbonyl groups for intermediates in the glycation synthetic pathway. Some of them may also exhibit free radical scavenging properties as aminoguanidine and pyridoxamine [14].
Another recent finding is that angiotensin-converting enzyme inhibitors (ACEi) are potent inhibitors of the formation of AGE. It has been postulated that ACE inhibition reduces the accumulation of AGEs in diabetes partly by increasing the production and secretion of AGERs into plasma. This increase in sRAGE could act as a mechanism to divert AGE from binding to the full-length RAGE receptor, acting as a “decoy” because this truncated form of the receptor has no downstream signaling capacity [15].
The aim of this study was to examine the contribution of AGEs in thioacetamide-induced liver fibrosis in rats and to assess the possible protective effect of AGE inhibitors, namely aminoguanidine as well as the angiotensin converting enzyme inhibitor; lisinopril.
Animals
Male Wistar albino rats (150-200 g) were obtained from the Animal House, Faculty of Medicine, Alexandria University. The animals were kept in a room under standard conditions of light, feeding and temperature. Animals used in this study were handled and treated in accordance with the strict guiding principles of the guide for care and use of laboratory animals of the Faculty of Medicine at Alexandria University. The experimental design and procedures were approved by the Ethical Committee of Faculty of Medicine -Alexandria University.
Experimental protocol
The animals were randomly divided into six groups, each of 8 animals as follows:
Group I (Normal control group): received physiological saline intraperitoneal (i.p) twice a week for 4 weeks.
Group II (Aminoguanidine control group): received aminoguanidine (Sigma-St. Louis, MO) in a dose of 100mg/ kg/d, i.p. [16] daily for 4 weeks starting from the first day of physiological saline administration
Group III (Lisinopril control group): received Lisinopril (Zestril- AstraZeneca Pharmaceuticals LP) in a dose of 10mg/ kg, i.p. [17] daily for 4 weeks starting from the first day of physiological saline administration.
Group IV (Thioacetamide (TAA) group): hepatic fibrosis was induced by i.p. injection of TAA (Sigma-St. Louis, MO) in a dose of 200 mg / kg twice a week for 4 weeks [18].
Group V (TAA-Aminoguanidine-treated group): received i.p. aminoguanidine (Sigma-St. Louis, MO) in a dose of 100mg/ kg, daily for 4 weeks starting from the first day of TAA administration.
Group VI (TAA- Lisinopril-treated group): received i.p. Lisinopril (Zestril-AstraZeneca Pharmaceuticals LP) in a dose of 10mg/ kg, daily for 4 weeks starting from the first day of TAA administration.
Collection of samples
At the end of the experimental period (4 weeks), rats were anaesthetized using thiopental sodium (40 mg/kg), blood samples were collected and serum separated for determination of AST and ALT activities [19].
Preparation of liver homogenate: Liver tissue was washed by cold normal saline solution, then it was homogenized in a homogenization buffer (0.05 M Tris-HCl pH 7.9, 25% glycerol, 0.1 mM EDTA, and 0.32 M (NH4)2SO4) containing a protease inhibitor tablet (Roche, Germany). The resulting solution was sonicated in an ice bath for 10 seconds followed by centrifugation at 13000 rpm, 4°C for 5 minutes. The supernatant was aliquoted and stored at -80°C and assayed for protein concentration using BCA kit (Pierce, Rockford, USA) using bovine serum albumin diluted in the lysis buffer as standard. The homogenate was used for the determination of:
Malondialdehyde(MDA) concentration: As a marker for oxidative stress, the amount of hepatic TBARS was measured by the thiobarbituric acid assay (TBA) as previously described by Buege and Aust [20]. MDA concentrations were calculated by the use of 1,3,3,3 tetra-ethoxypropane as a standard. The results were expressed as nmol/g wet tissue weight.
Reduced glutathione (GSH) concentration: Reduced glutathione was determined as previously described by Owens and Belcher [21] based on the reaction of 5, 5- dithiobis-(2-nitrobenzoic acid) (DTNB) with the GSH present. The results were expressed as μmol / g wet tissue weight.
Transforming growth factor-beta1 (TGF-β) concentration: Hepatic level of TGF-β was determined using an enzyme-linked immunosorbent assay (ELISA technique) [22].
Tissue inhibitor of matrix metalloproteinase (TIMP-1) concentration: TIMP-1 protein concentration in liver homogenate was determined with a TIMP-1 sandwich ELISA kit from Rand D Systems (Mannheim, Germany) [23].
Advanced glycation end products (AGE) concentration: AGE concentration in liver homogenate was determined with AGE ELISA kit, Roche Diagnostics (Mannheim, Germany) [24].
Hydroxyproline (HPO) concentration: Collagen concentration in liver homogenate was determined with HPO ELISA kit, Biosource [25].
Histological evaluation
Portions of the livers were processed for light microscopy. This processing consisted of fixing the specimens in a 5% neutral formol solution, embedding the specimens in paraffin, making 5μm thick sections, and staining the sections with hematoxylin and eosin and Masson’s Trichome staining.. The tissue slices were scanned and scored blindly. Pathological alterations consistent with fibrosis, enlargement, inflammatory infiltration, and breaking up of the hepatocellular limiting plates were observed in the portal tracts [26]. Liver sections were processed together for routine hematoxylin-eosin (HE) stain. Pathological diagnosis of each liver specimen was graded from 0 to VI according to the criteria described by Wang et al [27] (Table 1).
Statistical analysis: The results were expressed as mean values ± S.E.M. The data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparisons test as post hoc test. P<0 .05 was considered statistically significant.
Biochemical results
TAA administration resulted in a significant increase in serum AST and ALT activities as well as hepatic AGEs, MDA, TGF-β1, TIMP and HPO concentrations together with a significant reduction in GSH concentration in TAA group as compared to normal. Treatment with either aminoguanidine or lisinopril resulted in a significant decrease in serum AST, ALT activities and in hepatic AGEs, MDA, TGF-β1, TIMP and HPO concentrations as well as a significant increase in GSH concentration as compared to non-treated TAA group. No significant difference was found between untreated normal group and aminoguanidine-treated as well as lisinopril-treated control groups (Table 1).
| Degree of fibrosis | Score |
|---|---|
| No fibrosis | 0 |
| Slight fibrosis expanding to some portal areas and central veins | 1 |
| Marked fibrosis expansion, but without portal to portal bridging | 2 |
| Fibrosis expanding to most portal areas with occasional portal to portal bridging | 3 |
| Pseudolobules formed and partly replacing the normal architecture of the liver lobules | 4 |
| Occasional pseudolobules formed (incomplete cirrhosis) | 5 |
| Congested with pseudolobules, and between pseudolobules wide hyperplastic collagen fiber existed (complete cirrhosis) | 6 |
Table 1: Scoring system assessing the degree of fibrosis.
Histopathological results
Administration of TAA progressively induced several histological markers of cell death and liver fibrosis. In this regard, the tissue sections of untreated TAA animals showed numerous signs of portal and periportal hepatitis. Centrilobular degeneration of hepatocytes, in which hepatocytes appeared swollen with faint staining cytoplasm and some hepatocytes appeared unnucleated. Sinusoids in between appeared obliterated. Both aminoguanidine and lisinopril administration significantly reduced the histological signs of cell damage (Figure 1).
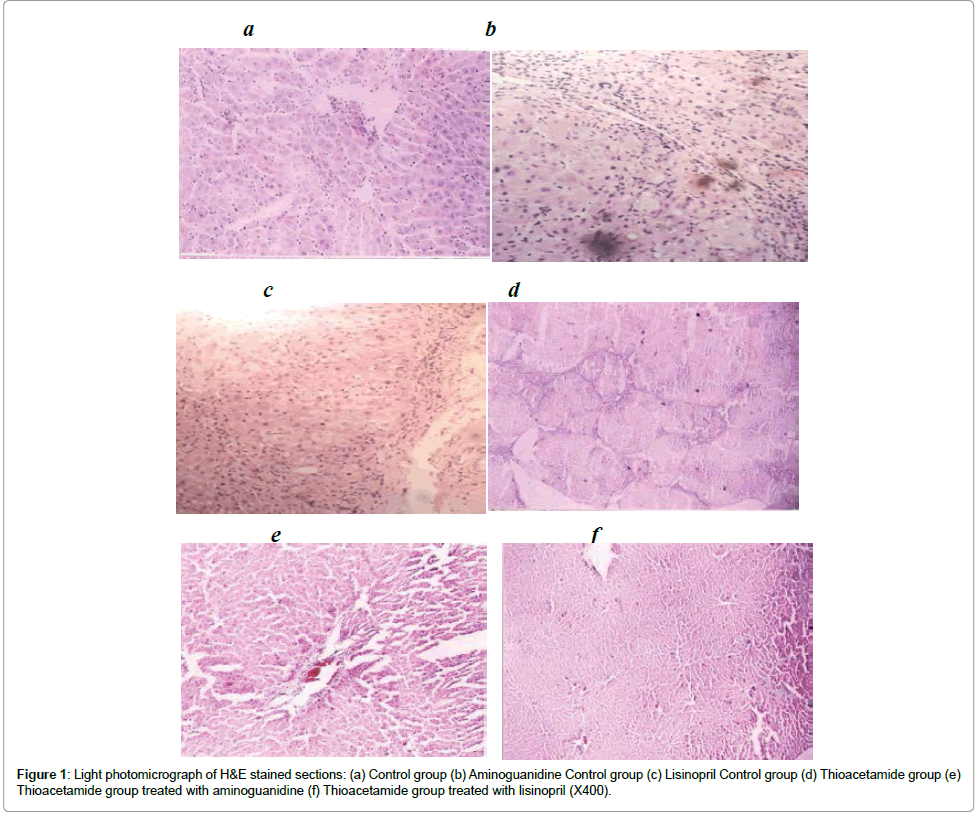
Figure 1: Light photomicrograph of H&E stained sections: (a) Control group (b) Aminoguanidine Control group (c) Lisinopril Control group (d) Thioacetamide group (e) Thioacetamide group treated with aminoguanidine (f) Thioacetamide group treated with lisinopril (X400).
Histopathological stages of the normal control as well as aminoguanidine and lisinopril control groups were all determined to be stage 0. TAA administration resulted in hyperplasia of connective tissues, but most of the histopathological stages were not above III. Aminoguanidine as well as Lisinopril-treated TAA rats resulted in obvious regression of fibrosis score (Table 2).
| Group | Hepatic fibrosis score | Serum ALT (U/L) | Serum AST (U/L) | Hepatic AGE (U/ m g protein) | Hepatic MDA (nmol/g wet tissue) | Hepatic TGF- b 1 (ng/ml) | Hepatic GSH (mg/g/g wet tissue) | Hepatic TIMP (ng/g wet liver) | Hepatic HPO ( m g /g wet liver) |
|---|---|---|---|---|---|---|---|---|---|
| Normal control | 0 ± 0 | 48 ± 15 | 85 ± 17 | 0.07 ± 0.02 | 18.31 ± 4.66 | 367.2 ± 14.6 | 124.2 ± 10.39 | 10.46 ± 2.6 | 42.4 6 ± 14.1 |
| Aminoguanidine control | 0 ± 0 | 51 ± 13 | 90 ± 12 | 0.09 ± 0.06 | 18.84 ± 5.10 | 389.7 ± 12.8 | 126.3 ± 15.12 | 10.79 ± 1.0 | 39.95 ± 10.21 |
| Lisinopril control | 0 ± 0 | 53 ± 11 | 89 ± 16 | 0.08 ± 0.04 | 19.09 ± 4.09 | 360 ± 14.7 | 127.7 ± 19.34 | 11.15 ± 2.7 | 41.76 ± 11.32 |
| TAA | 3.5 ± 0.2 | 137 ± 40* | 312 ± 76* | 0.30 ± 0.07* | 96.25 ± 9.63* | 1000 ± 28.4* | 73.9 ± 9.62* | 20.76 ± 5.7* | 288.46 ± 76.5* |
| TAA-Aminoguanidine | 1.5 ± 0.1 | 69 ± 10& | 103 ± 5 7& | 0.17 ± 0.05& | 28.29 ± 5.15& | 629 ± 18.3& | 111.5 ± 22.13& | 14.71 ± 3.9& | 79.96 ± 10.13& |
| TAA-Lisinopril | 1.4 ± 0.1 | 75 ± 16& | 110 ± 37& | 0.15 ± 0.06& | 25.85 ± 6.20& | 585 ± 21.4& | 113.2 ± 19.5& | 13.53 ± 4.5& | 89.31 ± 9.15& |
| F value | 78.83 | 66.95 | 57.96 | 39.16 | 42.73 | 82.52 | 43.28 | 52.76 | 75.92 |
| P | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
n: Number of rats in each group
*Significant compared to normal control group
&Significant compared to TAA group
Table 2: Hepatic fibrosis score, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities and hepatic advanced glycation end products (AGEs), malondialdehyde (MDA),transforming growth factor beta-1 (TGFβ1), reduced glutathione (GSH), tissue inhibitor of matrix metalloproteinase (TIMP) and hydroxyproline (HPO) concentrations (Mean ± S.E.M.) of the studied groups, four weeks after thioacetamide administration in rats.
In the present study, a significant increase in AGEs could be detected in TAA-induced liver fibrosis supporting the notion that these products might play a role in the pathogenesis of HSC activation and fibrosis.
AGEs are a heterogeneous group of molecules, formed in vivo both by non-oxidative and oxidative reactions of sugars and their adducts to proteins and lipids. It is now well established that formation and accumulation of AGEs progress during normal aging, and at an extremely accelerated rate under certain disease conditions, thus being implicated in various types of AGEs-related disorders such as diabetic vascular complications, neurodegenerative diseases ,cancers and fibrosis [28]. There is a growing body of evidence that activation of RAGE (receptor for AGEs) system is also implicated in these devastating disorders. Indeed, the engagement of RAGE with AGEs is shown to elicit oxidative stress generation and subsequently evoke inflammatory responses in various types of cells [29].
Liver is not only a target organ, but also an important site for clearance and catabolism of circulating AGEs. Although there are several studies to suggest the involvement of AGEs-RAGE system in various types of liver diseases such as non-alcoholic steatohepatitis, liver cirrhosis and cancers [30], as far as we know, there are no studies that have tested the effect of AGE inhibitors in liver fibrosis. Therefore, in this study, we investigated the pathological role of AGEs and the possible protective role of AGE inhibitors in TAA-induced liver fibrosis.
Enhanced RAGE expression in hepatic fibrogenesis has been previously shown in rats with cirrhosis induced by BDL and TAA treatment [31]. Our results are also in line with prior findings in CCl4-induced hepatic fibrosis where RAGE transcript and protein levels were upregulated until 6 weeks after the completion of CCl4 treatment [32]. Furthermore, it has been shown that α-SMA-positive HSC/myofibroblasts of the septal or portal interface, representing the prominent fibrogenic effectors, expressed RAGE in both fibrosis models.
AGEs have been reported to induce HSC proliferation by inducing cell proliferation and expression of genes relevant to HSC activation [33].
HSCs have been recently shown to express five advanced glycation end product receptors: Galectin-3, CD36, SR-AI, SR-BI and RAGE. All receptors, except SR-BI, showed up-regulation during HSC activation [34].
Indeed, a recent study conducted by Cai et al [35] demonstrated that specific targeting of RAGE using siRNA may inhibit RAGE gene expression effectively in the rat hepatic fibrosis model and attenuate the progression of established hepatic fibrosis. This therapeutic effect may be mediated via inhibition of the expression of NF-κB. These findings suggest that RAGE may be a new target to prevent hepatic fibrosis.
Another recent study investigated the effect of specific siRNAs targeting RAGE on the development of hepatic fibrosis (HF), using primary rat HSCs, which were isolated and cultured in vitro. The expression levels of RAGE together with IL‑6, TNF‑α, TGF‑β1, connective tissue growth factor, laminin, hyaluronic acid and N‑terminal procollagen III in the treated primary HSCs were significantly downregulated compared with those in the untreated. Thus, it can be deduced that RAGE‑specific siRNAs inhibited the expression of RAGE in primary rat HSCs and inhibited the development of HF [36].
The current study demonstrated an increase in hepatic MDA concentration in TAA-induced liver fibrosis and this might be a possible mechanism whereby AGEs induce liver fibrosis. Our results support the findings of Guimarães et al [34] who demonstrated that AGEs induce reactive oxygen species generation in HSCs and they concluded that this unveils a potential new route through which AGEs induce liver fibrosis in the metabolic syndrome.
Our results also demonstrated the possible mechanistic role by which AGE inhibitors may provide protection against liver fibrosis. This is possibly achieved by acting upon many possible sites in the proposed pathway; first by preventing oxidative stress, second by inhibiting the synthesis of proinflammatory cytokines (TGF-b) and thirdly by combating extracellular matrix accumulation via upregulating TIMPs. Previous results have reported the ability of AGEs to induce oxidative stress as well as proinflammatory cytokines [34] and the ability of aminoguanidine to inhibit oxidative stress [35] and to decrease lung fibrosis [36].
It is possible that the high TIMP-1 activity in the liver microenvironment prevents apoptosis of HSC and thus contributes to the fibrogenic progress on liver injury. Alternatively, it has been shown that TIMP can act as a transcription factor [37].
Our study also confirmed a growing evidence of cross-talk between the renin angiotensin system (RAS) and AGEs pathways [38], where the current study demonstrated that lisinopril suppressed the level of AGEs and resulted in a significant decrease in TGF-b, MDA and hydroxyproline levels as well as a significant increase in hepatic GSH and TIMPs. These results are in accordance with other results reporting an inhibitory effect of drugs targeting RAS on AGEs [39].
The mechanism(s) by which AGEs activate RAS might be through the RAGE-PI3-K/Akt-dependent pathway [40]. Conversely, angiotensin II can stimulate AGEs and upregulate RAGE expression [41]. Indeed, other studies reported the ability of the angiotensin receptor blocker; telmisartan, to inhibit AGEs production [42-46].
Our study opens new areas of therapy and provides new modalities for the management of liver injury. Targeting AGEs could represent a therapeutic option for patients at risk of developing liver fibrosis. Patients with hepatitis B or C infections, patients with autoimmune hepatitis, patients with nonalcoholic steatohepatitis due to obesity or type 2 diabetes, and perhaps elective transplant recipients might be candidates.