Journal of Developing Drugs
Open Access
ISSN: 2329-6631
ISSN: 2329-6631
Review Article - (2013) Volume 2, Issue 3
Aqueous solubility is one of the most influencing factors when it comes to bioavailability of the drugs. It is the main key for drug effectiveness and enhancing water solubility, which poses one of the greatest challenges in the pharmaceutical industry. Nearly half of newly developed drugs turn out to be insoluble or poorly soluble during initial screening process. Poor solubility limits drug delivery and formulation development. Various techniques have been developed to enhance solubility of poorly soluble drugs. Pharmaceuticals falling under the Biopharmaceutics Classification System (BCS) class II and IV are the main emphasis of this review as these drugs are of low solubility. The methods to improve drug solubility; solid dispersions, cyclodextrin complexation, dendrimers, nano-suspensions, co-solvency, pH adjustment, self-emulsifying drug delivery system, hydrotrophy, cocrystallization and ionic liquid formation are discussed.
<Keywords: Solubility; Biopharmaceutics Classification System (BCS); Solid dispersions; Complexation; Self-Emulsifying Drug Delivery System (SEDDS); Co crystallization; Ionic liquid formation
Scientists have been trying for decades to find the perfect way to increase the bioavailability of drugs by improving the solubility. A large number (~40%) [1] of New Chemical Entities (NCE) synthesized both in academia and industry suffer major setback due to poor water solubility [2]. This is due to the fact that the active pharmaceutical ingredient (API) needs to be in the solution in aqueous intestinal fluid that allows its partitioning across epithelial cell membrane (passive diffusion) [3].
Drug absorption relies heavily on two factors; solubility and permeability. As the number of poorly soluble drugs has increased, efforts to enhance solubility have evolved. The Biopharmaceutics Classification System [4] is used to categorize drugs based on solubility and permeability for formulation development. BCS consists of four classes based on solubility and permeability. Both BCS II and IV compounds are of low solubility (Table 1), and class IV compounds suffer from additional poor permeability issue. The solubility class boundary is based on the highest dose strength of a drug product. A drug substance is considered highly soluble when the highest dose strength is soluble in 250 ml or less of aqueous media over the pH range of 1-7.5 at 37°C [5]. The volume estimate of 250mL is derived from typical bioequivalence study protocols that prescribe administration of a drug product to fasting human volunteers with an eight ounce glass of water. A study published in 2006 suggests that out of the top 200 oral drug products in the United States; 31% are in class I, 32% are in class II, 23% are in class III, 6% are in class IV, and 11% are unclassified [6] (Table 1).
| Class I | Class II |
|---|---|
| High solubility High permeability |
Low solubility High permeability |
| Class III | Class IV |
| High solubility Low permeability |
Low solubility Low permeability |
Table 1: Biopharmaceutics Classification System (BCS).
The purpose of this review is to discuss few of the most commonly employed and two new techniques employed both in industry and academia for enhancing solubility for the convenience of pharmaceutical drug developers. Each technique is unique in its own way and works differently for each solute. This review will discuss the following techniques (or methods): solid dispersions, cyclodextrin complexation, dendrimers, nano-suspensions, co-solvency, pH adjustment, self- emulsifying drug delivery system, hydrotrophy, cocrystallization and ionic liquid formation.
The Noyes-Whitney equation provides indication on how the dissolution rate can be used to improve the solubility of poorly soluble drugs. By increasing the surface area and decreasing the particle size of the compound, dissolution can occur. Also, a greater rate of dissolution will happen by increasing the wettability of the surface compound and by decreasing its’ thickness. Decreasing the particle size of the will result in an increase of surface area [7].
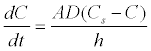
Where dC/dt is the rate of dissolution, A is the surface area available for dissolution, D is the diffusion coefficient of the compound, Cs is the solubility of the compound in the dissolution medium, C is the concentration of drug in the medium at time t and h is the thickness of the diffusion boundary layer adjacent to the surface of the dissolving compound [7]
Solid dispersion techniques can produce eutectic or solid solution products. A eutectic system is a mixture of chemical compounds, which solidifies at a lower temperature than any of the individual compounds. The eutectic melting point is sharply defined and occurs in a narrow range of temperatures. A eutectic system has a large surface area, which will enhance the dissolution. Solid solutions are analogous to liquid solutions that contain one phase. Solid solutions of a poorly water soluble drug dissolved in a carrier that has relatively good aqueous solubility, results in improved bioavailability [7]. Solid dispersions can be prepared by several procedures, such as fusion melt and the solvent methods [8]. Methods of carriers include emulsifiers, the use of polyethylene glycol, the use of Poly Vinyl Pyrrolidone (PVP), and others.
There are several disadvantages and advantages to the use of solid dispersions. Three disadvantages are the difficulty to scale-up the manufacturing method, the physical instability of the dispersion, and the large amount of carrier needed to facilitate an increase of the required release rate [9]. Two advantages are the chemicals needed for solid dispersions are already widely used in the pharmaceutical industry, so no extra toxicity studies are needed. Also, these approaches have a greater percentage than others of success in increasing solubility and the release rate. Solid dispersions are not commonly used because of the manufacturing, stability, and scale-up issues [9]. Examples of drugs that use the solid dispersion method are; nelfinavir mesylate (Viracept®, Agouron), ritonavir (Norvir®, Abbot), amprenavir (Agenerase®, Glaxosmithkline), calcitriol (Rocaltrol®, Roche), etravirine (Intelence®, Tibotec), and indomethacin (Indomethacin®, Eisai Co) [10].
Cyclodextrin (CD) complexation is another technique that helps to improve solubility. CDs have a specific cylindrical shape with a hydrophilic exterior, due to the presence of hydroxyl radicals, and a hydrophobic cavity of a specific size [11]. From this structure, cyclodextrins are slightly soluble in water. There are three types of cyclodextrins; alpha, beta, and gamma. The alpha has six glucose units, the beta has seven glucose units and the gamma has eight glucose units (Figure 1). Alpha cyclodextrin forms inclusion complexes with both aliphatic hydrocarbons and gases, such as carbon dioxide [8]. Beta cyclodextrin typically forms complexes with small aromatic molecules. Gamma cyclodextrin can accept more bulky compounds. Cyclodextrin incorporates lipophilic drugs inside its cavity by various interactions that in turn increase drug solubility and stability. The resulting complex still has hydrophilic hydroxyl groups exposed to the aqueous environment and the result is a water soluble CD-drug complex. Drugs with solubility in the micromole/liter range generally show greater solubility enhancement with cyclodextrins than with drugs who have solubility higher than the micromole/liter range. Low molar substitutions of cyclodextrin derivatives are better solubilizers than the same type of derivatives at higher molar substitutions [12].

Figure 1: Structure of various cyclodextrins.
An advantage to this method of enhancing solubilization by complexation is that it is achieved through specific interaction rather than changes in the bulk solvent properties. Unlike co-solvents, pH adjustments, and emulsion solubilizing systems, the dissociation is very rapid and quantitative which makes it predictable. An added significant advantage is that commonly used complexating agents like hydroxyl propyl beta cyclodextrin are not as toxic compared to other solubilizing agents such as surfactants [8]. A major disadvantage of the use of cyclodextrins is for formulating the ionizable drugs. Although, drug-CD complex is formed, the stability of the complex is greater in un-ionizable form; this was noted specially with chlorpromazine. The un-ionizable form has stability four times larger than the ionizable form. In order to enhance the solubility of ionizable drugs, pH adjustments are made. Beta-cyclodextrin will increase stability at lower pH [11]. This will help increase the solubility of some ionizable drugs. Also, the least amount of cyclodextrins should be used because if the particle becomes too large, reduced drug bioavailability and loss of preservative efficacy can occur. Drug solubility should be determined in the final formulation and under normal production conditions to be able to determine if too much, or too little, cyclodextrin is being used [12]. Few examples of cyclodextrin complexed formulations are beta-CD with Hydrocortisone, beta-cCD Paclitaxel (Taxol), beta-CD Ibuprofen, beta-CD Naproxen, and beta-CD Hydrochlorothiazide [11,12] (Figure 1).
Use of dendrimers is another important and well explored method used for drug solubility enhancement. Dendrimers have unique structures and properties, which spawn interest. There are several factors that affect dendrimer’s ability to enhance the drug solubility such as; generation size, concentration, pH, core, and terminal functionality [13].
Dendrimers are mainly used for enhancing the solubility of hydrophobic drugs. The main characteristic of dendrimers that favors them is their static micelle properties. These properties differ from orthodox micelle, as they do not require Critical Micelle Concentration (CMC) to assemble in the micellar form. Studies have shown that solubility enhancement with dendrimers are significantly superior to that of micelles and cyclodextrins. One article summarized various studies and found that generation number, pH of medium, size of dendritic micro cavities, and dendritic architecture are the factors that influence the efficiency of dendrimers as solubilizing agent [13]. Various suggested mechanisms to explain dendrimer’s solubilizing capabilities include, hydrophobic interactions, ionic interactions, internal encapsulation, covalent conjugation and hydrogen bonding.
One major advantage of dendrimers in drug delivery is that they provide an effective and resourceful polymeric design that could be cutomized for solubility enhancement [13,14]. Examples of drugs that use dendrimers are: PAMAM dendrimer in Nifedipine®, PEG polyether dendrimers in Indomethacin®, PEG-PAMAM dendrimer in Methotrexate®, and PEGylated lysine dendrimers in Artemether® [13].
Drugs can be made into nano-suspensions of particles with diameters less than 100 nanometers (nm) [15-18]. Nano- sized compounds are used to improve solubility of poorly-water-soluble compounds and are sub-micron sized colloidal dispersions of pure drug particles in an outer liquid phase. An added advantage when using nano-suspensions to enhance solubility is this application allows for increased solubility and dissolution rates of the compound. An increase in the dissolution rate can occur because of the increase in surface area.
There are various approaches (precipitation, microemulsion, high-pressure homogenization and milling methods) to create nanosuspensions, two of the most common ones; solvent diffusion method and melt emulsification methods are discussed. In the solvent diffusion method, a solvent-in-water emulsion is formed. This includes a water miscible solvent with the dissolved drug in the dispersed phase. The selection of the solvent and stabilizer are very important in creation of nano-size particles. The ideal combination of solvent and stabilizer is one that forms a stable emulsion. The final step is the homogenization process that dilutes the emulsion with vigorous mixing. The melt emulsification method starts with a hot emulsion with the melted drug in the dispersed phase. The emulsion is formed from homogenization and cooled to solidify the droplets of the melted drug. A disadvantage of this method is that it produces much larger particles [19]. Nanosuspensions have been used to formulate various drugs and a few examples are amphotericin B, tarazepide, atovaquone, paclitaxel, and bupravaquon.
Addition of a water miscible solvent can be used to make a drug water-soluble. This procedure is simple to produce and evaluate. The best drug candidates for co-solvent method are compounds that are lipophilic or compounds with a high crystalline structure that are highly soluble in the selection solvent mixture. By using co-solvents, a compound’s aqueous solubility can be increased quite significantly than just the compound alone [8]. This is achieved by first dissolving the compound in a non-aqueous but water miscible solvent and then adding water sufficiently to maintain the drug solubility and adjusting the dose.
Another advantage to co-solvency method is that high concentrations of the compound can be dissolved compared to other methods. The co-solvent method may be paired with other solubilization techniques such as pH-adjustment to increase solubility. Some disadvantages of this method are the chemical stability of the insoluble drug is worse than in the crystalline state, as with all solubilized forms. The toxicity and tolerability of the excipients must be closely watched. Also, for intravenous administration, uncontrolled amorphous or crystalline precipitation may occur upon dilution with aqueous media. This occurs because the drug is insoluble in water and after precipitation forms the co-solvent mixture is not readily able to re-dissolve. Embolism and local adverse effects are latent risks at the injection site. Examples of co-solvent products are Nimodipine Intravenous Injection (Nimotop®, Bayer) and Digoxin Elixir Pediatric (Lanoxin®, GSK) [8].
The modification of pH (via buffering) is a good option for ionizable drugs. Ionizable drugs can be protonated if have basic groups (e.g., amine) or deprotonated if acid group (e.g., carboxylic acid) is present. Excipients are also used to increase environmental pH in tablet or capsule dosage form. Adjustment of the pH is frequently combined with co-solvents for further increase in solubility [10]. The pH-adjusted formulations are simple to produce and development progresses quickly. Some disadvantages of this method are tolerability and toxicity may occur due the use of non-physiological pH’s. The drug molecules in a pH-adjusted formulation could also become less soluble and precipitate upon dilution in aqueous media and if were administered intravenously, could cause emboli. Moreover, since the drug is less stable chemically in an aqueous environment than in crystalline form, the employed pH may enhance the hydrolysis or catalyze other degradation mechanisms [8]. The pH adjustment method has been used to formulate commercially and examples of available products are; phenytoin injection (Epanutin® ready mixed, Pfizer) 50 mg/ml with propylene glycol 40% and ethanol 10% (1.1 mmol Na+ per 5 ml ampoule) [20].
Hydrotrophy can be defined as the ability of a compound (hydrotropes) to increase the aqueous solubility of another a sparingly soluble compound. In general, hydrotrophes are ionic organic salts such as sodium benzoate, urea, sodium salicylate, nicotinamide, sodium acetate and sodium citrate that are increase solubility of poorly soluble drugs [20]. This method does not require chemical modification, preparation of emulsion system, or use of organic solvents, which are the advantages of this method [8].
In hydrotrophy process, a large amount of a second solute is added to the first solute in order to make the second solute more soluble. The interactions between the hydrotropic agent and the solute are weak. Disadvantages of this method are that hydrotropes may self-assemble in solution and lose the ability to enhance drug’s water solubility. Some advantages of hydrotrophy are; the mixing step is done only with the drug and hydrotrope in water, has high selectivity and does not involve emulsification [8]. Some examples of drugs where hydrotropes have been used are procaine/HCl in Riboflavin®, absorbic acid in Saquinavir®, sodium salt of Ibuprofen in Ibuprofen®, sodium benzoate in Carbamazepine®, and sodium salicylate in Paracetamol® [20].
Self-Emulsifying Drug Delivery Systems (SEDDS) are involved in lipid formulations. These formulations consist of isotropic mixtures of drugs, which are commonly lipids or surfactants [21-24] with one or more hydrophilic co-solvents or co-emulsifiers. This system forms an emulsion instantly after slight agitation and dilution with water. These emulsions produced are a droplet size extending from a few nanometers to numerous microns. This system can be used with all BCS class drugs to help improve their solubility. SEDDS helps maintain solubility in the gastrointestinal tract by avoiding the dissolution step, which can limit the absorption rate of hydrophobic drugs. The two main factors that affect the release rate of the drug in SEDDS are the particle size and the polarity of the droplets. For the most effective formulation, it is best to keep the number of excipients to a minimum. Excipients are the backbone of SEDDS. The most frequently used excipients are lipids, surfactants and co-solvents. The best choices of excipient are those that increase drug solubility. Lipids are good for solubilizing lipophilic drugs and enhancing the transportation of lipophilic drugs [25].
Surfactants are ampiphilic, so they are able to dissolve large amounts of hydrophobic drug compounds. Co-solvents typically use high concentrations of hydrophilic surfactants. A mixture of surfactant and co-surfactant leads to formation of Self-Micro Emulsifying Drug Delivery System (SMEDDS), which ranges from droplet size between 100 and 200 nm [26].
An advantage with the use of SEDDS and SMEDDS is that tablets and capsules can be developed while sustaining good flowability, cohesive properties, and good content uniformity. This method allows excellent product design, performance, and manufacturability. It has not been until recently that understanding in detail the use of SEDDS and SMEDDS on drug deposition as lipid formulations. A major disadvantage is that this method needs further examination to identify a wide range of self-emulsifying formulations that can help with delivery of poorly soluble drugs. SEDDS have been used by drugs such as isotretinoin in Accutane® (Roche), cyclosporine A in Gengraf® (Abbott), ritonavir in Norvir® (Abbott), cyclosporine in Panimum bioral® (Panacea Biotec), and amprenavir in Agenerase® (GSK) [10].
The cocrystal formation via crystal engineering approach requires a library of cocrystallizing agents or coformers [27]. A pharmaceutically acceptable, nontoxic coformer must be chosen to result in a pharmaceutically acceptable cocrystal. This limits the coformer pool to those molecules that have been previously declared safe for human consumption. They can be selected from the collections, such as Generally Recognized as Safe (GRAS) by the U.S. Department of Health and Human Services and Everything Added to Food in the United States (EAFUS) [28-30].
A popular approach to cocrystal design is the consideration of pKa [31]. Generally, salts and cocrystals differ from each other by absence of proton transfer in co-crystals. It has been suggested that a pKa difference of minimum two units (between an acid and a base) is required to form a salt [32]. Also, it was noted for the system of acid and base with similar pKa, the proton transfer in the solid state may not be predictable and existence of a continuum has been suggested [33]. In cases where it is not known whether the molecules in the new solid state are held together by hydrogen bonding or by electrostatic interactions (after the proton transfer), assigning a characteristic name (cocrystal or ionic solid) is not feasible.
Shape complementarily or the structural fit was observed to be the dominant factor in cocrystallisation of cis-itraconazole with a series of 1,4-dicarboxylic acids [31]. The cocrystallization did not take place between cis-itraconazole and maleic acid with Z regiochemistry about the C=C bond or from 1,3- or 1,5-dicarboxylic acids. Thus, geometrical requirements seem to be more important than acid-base strength match for successful a co crystallization reaction. Similar to polymorph screening, making cocrystals is an empirical exercise. However, likely hood can be increased by choosing synthons (coformers) such that there is highly likely of complementary hydrogen bond formation. Etter et al., proposed the guidelines that can be followed to facilitate the design of cocrystals or any hydrogen bonded solid [34].
There are several techniques can be used to synthesize cocrystals. The oldest is the evaporation of a solution containing stoichiometric amounts of the active pharmaceutical ingredient and the coformer. Recently, many new and efficient alternatives have been developed including, mechano chemical methods of neat [35], Liquid-Assisted Grinding (LAG) [36], thermal techniques [37,38] and the Sonic Slurry method [36,37]. It was reported that the chances of cocrystals formation in solvent mediated reaction is higher if saturated conditions are maintained for both the components, otherwise no cocrystals or mixture of products will result [39].
It has been demonstrated that cocrystallized API’s can exhibit an enhanced dissolution rate for BCS II compounds, translates into higher bioavailability. McNamara et al., prepared cocrystal of an API (2-[4-(4-chloro-2-fluorphenoxy) phenyl] pyrimidine-4-carboxamide) with glutaric acid. The Intrinsic dissolution of the compounds in the compressed disk form (both API and cocrystal) revealed cocrystal to be 18 times more soluble than the API. This cocrystal powder in capsule and the API when fed to the dogs at two dose levels (5 and 50 mg/kg), the cocrystal was found to be about 4 times more bioavailable than the API [40].
Very recently, Ionic Liquids (ILs) have found great applications in efficient and environmentally benign chemical processing and chemical analysis [41,42]. By definition the Ionic Liquids (ILs) are organic salts with Melting Points (MP) below 100°C or more often even lower than room temperature [43-47]. These compounds posses dual capability of dissolving both polar and nonpolar species and the most useful feature is that they do not evaporate even at high temperatures [48-51]. Most commonly, ILs are based on nitrogen-rich alkyl substituted heterocyclic cations, with a variety of anions (e.g., 1-ethyl-3-methylimidazolium tetrafluoroborate). Although, the reasons for low melting point of ILs are not clear, it is stated that, ILs consist of bulky inorganic anions with delocalized charged organic cations, which prevents the formation of a stable crystal lattice or random molecular packing resulting in lower melting points [52]. Due to these remarkable characteristics, ionic liquids have been used as, medium for liquid-liquid extractions [53-55], mobile phase additives in High Performance Liquid Chromatography (HPLC) [56,57], electrolytes in Capillary Electrophoresis (CE) [58-62], matrixes for Matrix Assisted Laser Desorption Ionization Time- Of-Flight Mass Spectrometry (MALDI-TOF MS) [63,64], stationary phases for Gas Chromatography (GC) [65-68] and as modifiers in Micellar Electro Kinetic Chromatography (MEKC) [69-71].
The physical form of a drug substance is of great importance as it directly affects the manner in which the material is formulated and presented to the consumer. It also influences more fundamental characteristics such as solubility and dissolution rate, which, in turn, affect bioavailability. These factors, as well as the patentability of new physical forms, have resulted in a flurry of activity in screening for novel solid forms, including salts, polymorphs, pseudo polymorphs (solvates), and co crystals [72]. It has been reported; more than 50% of drugs marketed are salts [73]. This allowed, exploring the effects of counter-ion modification on the pharmacological outcomes of the drug salt [74]. Very often, counter-ion choice is limited by the synthesis or purification routes, rather than rational choice or screening. Nevertheless, via well-established chemistry, counter-ions can be replaced. In case, when one of the ions is large enough to hamper crystal packing, result is an ionic liquid form of the salt.
Recently Rogers et al., have reported ionic liquids formed from active pharmaceutical ingredients (APIs) [75]. In case of a salt void of crystalline phase will not face the issues of polymorphic conversions. Additionally, ILs in general and especially, Room Temperature Ionic Liquids (RTILs), salts that are liquid phase at ambient temperature, are already liquid and can potentially absorb much faster to proved rapid onset of the drug. However, it has been observed, when an amorphous form of drug added to the water, could lead to sudden crystallization [76] due to reaching the saturation limit. Apart from the advantages of the liquid state, a second biologically active counter ion can be included which might lead to dual functional drug [77,78].
For BCS class II drugs, enhancing solubility would be very efficient for increasing bioavailability. Developments of newly synthesized compounds are frequently stopped because of solubility issues. Solubility can be enhanced by many techniques and each technique will increase certain drug solubility by a number of folds. The various techniques described above alone or in combination can be used to enhance the solubility of the drug. There are many other sources of applications used to enhance solubility. These applications are micellar solubilization, super critical fluid process, crystal modification, amorphization, spray freezing, use of surfactants, salt formation and several others. It is necessary to use one of these techniques and the ones mentioned above to increase the solubility of drugs because the bioavailability is affected by low solubility. Each technique has advantages and disadvantages, which is important when deciding the appropriate method for the drug selection. It is imperative that the correct technique is chosen in order to decrease the possibility of errors. A better understanding of how to increase the solubility of drugs with different methods has been developed by academic and industrial research and this science will lead to development of efficient formulation for poorly soluble drugs.