Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Review Article - (2012) Volume 1, Issue 2
The intervertebral disc (IVD) is a multi-component structure consisting of a heterogeneous population of cells that form the central nucleus pulposus, encased by the fibrous annulus fibrosus and the cartilage end-plate. The essential function of the IVD is to withstand biomechanical forces, confer tensile strength and flexibility in motion to the spine. Disc degenerative disease (DD) is a prevalent ailment that affects the general population, often manifesting either in the form of lower back pain or as deformities of the spine such as degenerative lumbar scoliosis or in severe cases as disc herniation. With the aid of mutant mouse models generated through traditional knock-out strategies and spontaneous mutants, scientists have been able to elucidate some of the fundamental mechanisms of embryonic IVD development. Mutual interaction between the notochord and vertebral bodies are instrumental in the proper formation of the IVD. In this review, the known and proposed molecular mechanisms underlying these processes and the areas that require further investigation are discussed. Sufficient knowledge on the molecular mechanisms of IVD formation and the etiology of IVD degeneration is currently lacking and this has greatly hampered efforts to design appropriate and effective therapies for DD. With the dawn of the next-generation sequencing and better tools to engineer the genome, elucidation of the mechanism of IVD formation and the molecular basis of the pathology of DD ought to be an appealing avenue for researchers to pursue.
<The vertebral column is the most crucial framework of all vertebrates, functioning to provide support, flexibility and protection of essential spinal nerves. It is comprised of the characteristic metameric arrangement of the vertebral bodies linked together by intervertebral discs (IVDs) [1]. An indispensable aspect of the vertebral design, the IVD serves to withstand biomechanical forces and confers tensile strength and flexibility in motion to the otherwise rigid spine [2]. Disc degeneration is a major cause of lower back pain prevalent among adults. It is a progressive disorder that worsens with age and may even lead to permanent disabilities. It also imposes a substantial socioeconomic burden on the individual as well as the health care system. The economic impact of lower back pain is striking, and is in excess of the costs of coronary artery disease and the total costs of stroke, respiratory infection, rheumatoid disease and diabetes combined. Direct medical costs in the USA annually exceed USD $30 billion [3-5].
The biochemical hallmarks of degenerative disc disease (DD) are decreased proteoglycan content of the IVD, which in turn reduces the water retention ability of the nucleus pulposus and a marked increase in the amount of collagen [6]. This significantly alters the IVD structure, thereby the load-bearing capacity of the affected spine [6-8]. Owing to the avascular nature of the IVD, its capacity for self-renewal or repair is poor [9]. Most of the current clinical therapies involve treating the symptoms with medication and physiotherapy rather than to solve the root of the problem – restoring the IVD to its native state and function. Surgeries such as arthrodesis are often performed as a last resort, and are known to involve complications such as adjacent level disease [1,6]. Recent developments in cell transplantation therapy for DD using bone marrow-derived adult stem cells, termed Mesenchymal Precursor Cells (MPCs), appears promising [10]. Clinical trials have just begun recently in 2012 at the Washington Center for Pain Management in US. However, the effectiveness and safety of the MPC transplantation remains to be assessed in human patients. All the while a lack of sufficient knowledge on the development of the IVD and the etiology of DD, have been the chief factors accountable for the delay in the creation of appropriate and effective therapies. While the MPC transplantation for DD might set the trend for the generation of more such therapies, we are still far from establishing a cure for DD. Therefore, understanding the embryonic IVD developmental process and its regulatory network are of utmost importance to gain a complete insight into the processes which malfunction in DD and identify areas for therapeutic intervention.
Elucidating IVD development is impossible in human embryos for ethical reasons. The mouse has long served as one of the excellent in vivo model systems to study the morphogenesis of IVD. Its largely conserved genetic background and vertebral structure, relatively short gestation period (19 days), a fully sequenced genome, easy availability and amenability for genetic engineering, as well as established gene manipulation techniques, are all determining factors in its usefulness for studying IVD [11,12]. The development of IVD will thus largely be discussed based upon the mouse model in this review.
Natural mouse mutants served as the starting point for the multitudes of mutant mouse lines that are now available in the public repository for researchers world-wide. Spontaneous mouse mutants with a hunchback, short or kinked-tails or a scoliotic backbone not only triggered our curiosity but also urged us towards a reverse genetics approach [13,14]. The generation of more such mutants through genetic engineering enabled us to understand the basis of such defects of the spine [15-18]. Similarly, lineage-tracing studies and in vivo imaging utilizing reporter genes have helped us to track and visualize specific cell types involved in the morphogenesis of embryonic IVD [19-22]. While such mutant mouse models along with studies on C.elegans, zebrafish, rat, rabbit, canine, sheep, bovine and human patient IVD samples provided us with a basic understanding of the morphogenesis and characteristics of the IVD, the exact molecular mechanisms of its development, homeostasis and degeneration are currently poorly defined.
The aim of this review is to provide a summary of the known and proposed molecular mechanism of embryonic IVD development identified in mouse models. We also propose the prospective direction that could be taken to tackle the existing deficits in understanding IVD formation.
Briefly, the mature IVD is a multi-component fibro cartilaginous structure consisting of a gelatinous central nucleus pulposus, encased by the fibrous annulus fibrosus, which in turn is sandwiched between rostrally and caudally positioned cartilaginous endplates [1]. The distinct biochemical properties of each of these IVD components are critical to execute their unique biomechanical functions. For instance, the semi-fluid nature of the nucleus pulposus enables it to act as a shock absorber. It helps to withstand the compressive forces acting on the spine. Indeed, alterations in the gelatinous consistency of the nucleus pulposus and its conversion to a more fibrous form have been attributed to the loss of notochordal cells or large vacuolated cells and an increase in chondrocyte-like cells within the degenerate IVD of adults. The more fibrous form of the nucleus pulposus diminishes its load-bearing ability [23,24]. Similarly, the fibrous nature of the annulus fibrosus enables it to endure tension as well as to hold the central nucleus pulposus in place during compression. The cartilage end-plate, a thin layer of hyaline cartilage, mainly acts to provide continuity by linking the adjacent bony vertebrae to the annulus fibrosus. Together, these components are thus able to transmit and evenly distribute the load from the body weight and general activity [1].
Studies on the various animal models have shown that the nucleus pulposus is entirely derived from the notochord, whereas the annulus fibrosus and cartilage end-plate are sclerotome-derived structures [2]. The distinct morphological and structural characteristics of the annulus fibrosus and the cartilage end-plate tissues despite their shared cellular ancestry indicate that crucial molecular mechanisms underscore their specification during embryonic development. Investigation of the numerous transcription factors (TFs) by elucidating the gene regulatory network (GRN) involved in embryonic IVD morphogenesis will provide clues to the exact cell-type specification mechanism involved, which may greatly assist in the refinement of cell therapy for DD.
Like in all other developmental programs, formation of the embryonic IVD is a highly coordinated multi-step process, beginning with patterning, specification and morphogenesis of the tissues before differentiation takes place. IVD development involves a concerted action of intrinsic instructions as well as external signals and cues from the notochord and its surrounding vertebral bodies (VB). The formation of IVDs is thus a result of the mutual development of the notochord and the VB. Perturbations at any of the key stages can give rise to an abnormal IVD owing to disrupted downstream processes. In the following sections, the embryonic IVD development commencing with the formation of notochord and sclerotome will be discussed. The molecular mechanisms involved and the hypothesized processes by which components of the IVD are formed will be reviewed as well. A summary of the genes involved in IVD genesis and the corresponding mouse mutants that revealed the mechanisms are provided in Table 1.
| Developmental process | Function | Gene(s) involved | References for corresponding mouse models |
|---|---|---|---|
| Notochord formation | Initiation | Foxa2 | [28-29] |
| Notochord formation | Formation (rostral) | T | [13] |
| Notochord formation | Formation (caudal) | Noto | [27] |
| Notochord Maintenance | Viability | c-Jun | [40] |
| Notochord Maintenance | Viability | Sox5, Sox6, Sox9 | [16-17] |
| Notochord Maintenance | Proliferation | Shh (signaling) | [25,44,64] |
| Notochord Maintenance | Viability & proliferation | Tead1 & Tead2 | [46] |
| Notochord Maintenance | Viability &/or proliferation | Sd mutant; unknown gene | [14] |
| Notochord sheath | Formation | Sox5 & Sox6 | [16] |
| Notochord sheath | Formation | Shh (signaling) | [25,44,64] |
| Notochord sheath | Formation | Sd mutant; unknown gene | [14] |
| Sclerotome | Specification | Shh (signaling) | [25,44,64] |
| Sclerotome | Specification | Smo | [44,62] |
| Sclerotome | Specification | Gli2 & Gli3 | [63] |
| Sclerotome | Specification | Nog & Grem1 | [61] |
| Sclerotome | Specification | Pax1 & Pax9 | [48] |
| Sclerotome | Specification | Mfh1 | [18,59] |
| Sclerotome | Proliferation | Pax1 & Pax9 | [48] |
| Sclerotome | Proliferation | Mfh1 | [18,59] |
| Sclerotome | Differentiation | Meox1 & Meox2 | [66] |
| Sclerotome | Differentiation | Nkx3.2 and Nkx3.1 | [15,49] |
| Sclerotome | Differentiation (AF fate) | Mfh1 | [18,59] |
| Sclerotome | Differentiation (AF fate) | Tgfbr2 (Tgfb-signaling pathway) | [69] |
| IVD anlagen | Maintain boundary | Tgfbr2 (Tgfb-signaling pathway) | [69] |
| NP morphogenesis | Reorganization of notochord | Col2a1 | [72] |
| NP morphogenesis | Reorganization of notochord | Shh (signaling) | [44] |
| NP morphogenesis | Reorganization of notochord | Tgfbr2 (Tgfb-signaling pathway) | [69] |
| NP morphogenesis | Reorganization of notochord | Pax1 & Pax9 | [48] |
Table 1: List of genes or gene locus involved in intervertebral disc development.
Notochord formation
The foundation for the IVD is first laid at one of the very early stages of embryonic development – the node formation. At the embryonic level, the organizer node is formed at E7.5, which gives rise to the presumptive notochord cells of only trunk and the tail region, while the anterior head process notochord is derived from non-node cells [19]. The notochord is a crucial signaling center essential for the dorsalventral (D-V) patterning of the surrounding paraxial mesoderm that will give rise to the sclerotome and subsequently the annulus fibrosus and cartilage end-plate of the IVD. Moreover, the notochord cells themselves are the precursors of large-vacuolated cells in the nucleus pulposus of the mature IVD [20,25]. Perturbation of notochord formation will thus result in a hypoplastic or dysmorphic IVD.
Node-derived notochord specification and differentiation is a Forkhead box A2 (Foxa2)-, Brachyury (T) - and Notochord homolog (Xenopus laevis) (Noto) - dependent process. While formation of trunk notochord and caudal notochord are dependent on T [13,26] and Noto [27], Foxa2 (a forkhead box TF) is absolutely crucial for notochord formation, lack thereof results in a failure of notochord initiation as its targeted deletion in mouse resulted in a complete lack of notochord and pre-natal lethality [28,29]. Investigations of E8.5 Foxa2 mutant embryos showed that the notochord formation, including the head process, was never initiated. Plus, inference from prior studies and microarray profiling by Tamplin et al. [30] have identified Noto, T and Sonic Hedgehog (Shh) to be downstream of Foxa2 in the node/ notochord formation [29]. More recently, by intersecting the data from ChIP-Sequencing for Foxa2 in liver tissue and microarray profiling of Noto+/eGFP notochord cells, the authors identified 9 novel direct binding targets of Foxa2 specific for notochord [31]. The functionality of these novel targets in the notochord needs to be assessed though.
Likewise, T (a T box TF) is essential for the differentiation of the notochord from the midline mesoderm [26,32]. The T-/- mutants are devoid of the trunk notochord (but retain the head process notochord) and die pre-natally by E10.5 because of allantois defect [13]. Even though extensive studies on T function and its downstream targets have been investigated in Ciona intestinalis [33], Danio rerio [34] and Xenopus laevis [35,38], the identification of direct targets in mouse has been carried out only recently. The authors used ChIP-Chip on mouse embryonic stem (ES) cells and identified components of the WNT and FGF signaling pathways to be regulated by T. Interestingly, Foxa2 is among those targets, indicating a possible regulatory loop [39]. It has to be noted, however, that this study was carried out in ES cells and is not tissue-specific.
Similar to T, Noto (previously known as Not; a homeobox TF) was identified through positional cloning of a spontaneous mouse mutant, the truncate (tc), which showed a lack of the caudal notochord [27]. Targeted mutation of Noto confirmed its role in the posterior notochord formation. Furthermore, Noto is postulated to be genetically downstream of Foxa2 and T in mouse owing to its complete absence of expression in either one of the null mutants [27,28].
While the early embryonic lethality in all of the Foxa2, T, and Noto mutants prevents further study of their molecular roles in subsequent IVD development, Foxa2-cre [21], mice with tamoxifen-inducible Crerecombinase expressed from Foxa2 locus [22] and Noto-cre [20] mouse lines can be used to overcome this issue and delineate their tissuespecific roles.
Notochord maintenance
Besides the three key regulators involved in notochord formation, several more are needed to maintain the notochord cell population. SRY-box containing gene 5 (Sox5), SRY-box containing gene 6 (Sox6), SRY-box containing gene 9 (Sox9), Jun oncogene (c-Jun), Shh and TEA domain family member 1 (Tead1) and TEA domain family member 2 (Tead2), regulate either the proliferation and/or apoptosis of the notochord cells.
The Sox trio (Sox5, Sox6 and Sox9) and c-Jun (major component of AP-1 TF complex) are essential for late notochord cell survival, but not needed for its proliferation or formation per se. The targeted knock-out of both Sox5 and Sox6 (Sox5-/-Sox6-/-) resulted in a loss of notochord cell population more dramatically in the rostral than caudal segment. In addition, the individual Sox5- or Sox6-null mutants do not possess the same notochord defects observed in the double null, demonstrating their redundancy in this function [16]. Similarly, upon conditional knock-out of Sox9 (in Ck-19 expressing cells), the notochord disintegrated gradually in a rostral-to-caudal progression and in the conditional knock-out of c-Jun (in Collagen, type II, alpha 1 (Col2a1)-expressing cells), a significant decrease in cell numbers in the IVD was observed at E13.5 [17,40].
While c-Jun, Sox5 and Sox6 and Sox9 are all essential for notochord survival, the Sox5 and Sox6 genes appear to play a relatively more important role. Notochord cells in the c-Jun- and Sox9-deficient mice survive long enough to differentiate into large vacuolated cells in the nucleus pulposus, unlike in theSox5-/-Sox6-/- mutants where no nucleus pulposus forms. The development of the inner annuli is also impaired in these Sox5-/-Sox6-/- mutants [16,17,40].
On the contrary, Shh signaling is essential for notochord cell proliferation. Known to be an activator of proliferation [41-43], analysis of the conditional knock-out of Smoothened (Smo) (a component of Shh signaling pathway) in Shh-expressing cells showed a marked reduction in notochord cell proliferation [44].
Notably, Shh and T, which are genes known to be expressed in the early notochord [44,45], were not affected by the loss of both Sox5 and Sox6, and Sox9 [16,17], suggesting that these genes could either be in a parallel pathway or upstream of Sox5, Sox6 and Sox9 in the genetic regulation of notochord maintenance.
Tead1 and Tead2 (Tead family TFs; contain the TEA DNA-binding domain) regulate both the proliferation and viability of the notochord cells. A considerable decrease in proliferation and increase in apoptosis was observed in general in the mesoderm of Tead1-/-Tead2-/- mutants [46].
Apart from these loss-of-function mutants, the Danforth’s Short tail (Sd) spontaneous mutant demonstrates a dramatic loss of notochord cells and a failure of notochord cell differentiation into large vacuolated cells that are characteristics of the nucleus pulposus [14]. Although the actual gene responsible for the Sd phenotype has yet to be identified, the mutation is believed to be of the gain-of-function type based on an enhancer-trap assay [47]. Accordingly, it is possible that the gene in Sd mutant might be directly or indirectly (through other genes) regulating the proliferation, apoptosis and differentiation of notochord cells. Identification of this gene and its downstream targets is thus essential to reveal its true molecular functions in IVD development.
The consequence of this overall decline in notochord cell numbers is the defective development of the nucleus pulposus. As a result, impairment of the IVD often manifests as a fused vertebrae and a scoliotic backbone in the adult mice.
Peri-notochord sheath formation
As the rod-like notochord is formed, a sheath of collagen fibrils, basal lamina and sulfated proteoglycans envelops the notochord cells. This process of peri-notochordal sheath formation is completed by E10.5 [44]. The exact function(s) of the sheath, the cells responsible for its synthesis and the molecular mechanisms underlying this process are currently unknown. The notochord sheath defect in numerous mouse mutants provides some clues to address these questions. Sox5-/-Sox6-/-, Sd and Shh-deficient mutants all possess sheath formation defects, indicating their importance in its development [14,16,44].
Firstly, it would not be unreasonable to postulate that the cells closest to the sheath would be responsible for its establishment. This could mean that either the notochord cells themselves or the surrounding sclerotomal cells or both are necessary for the secretion of the sheath components. When Shh is temporally removed (using tamoxifen-inducible Cre allele ShhcreERT2 and Shhf/f mouse lines) before sheath formation (E8.5), a rudimentary sheath was observed later (E11.5) in the rostral but not in the caudal portion. This correlated with the presence of a notochord, albeit thinner, in the rostral region but its complete absence from the lumbar region onwards [44]. Likewise, in the Sd mutants, the notochordal sheath is missing only in the regions lacking notochord cells [14]. Moreover, in the targeted knock-out paired-box (Pax1-/-Pax9 -/-) and NK3 homeobox (Nkx3.2-/- and Nkx3.1-/-Nkx3.2-/-) mouse mutants, where there is a significant loss of sclerotomal cells surrounding the notochord, but the notochordal cells are still present, the notochordal sheath appears intact [48,49]. These observations strongly argue for the role of notochord cells in sheath formation.
On the contrary, in the Sox5-/-Sox6 -/- mutants, sheath formation was never observed despite the presence of a reduced number of notochord cells. One might be tempted to conclude that the notochord cells are therefore not accountable for the sheath formation. However, it is also possible that the ability of these rudimentary notochord cells to secrete proteoglycans and collagens is impaired in the absence of the Sox genes. Indeed, Sox5, Sox6 and Sox9 are known to be direct activators of Aggrecan (Acan) and Col2a1 genes, which are key components of the notochord sheath [50-53]. Thus, these observations lend support to the hypothesis that notochord cells are responsible for sheath formation and that Sox5 and Sox6 may be the TFs critical for its initiation [16]. Besides, the fact that the notochord sheath is thinner in the rostral region of Shh-mutants indicates an incomplete/ abnormal sheath formation. Hence, as the authors proposed, Shh signaling remains essential for sheath formation [16,44]. Whether Sox5, Sox6 and Shh signaling are interconnected in this process has to be evaluated. Analysis of Sox5 and Sox6 expression in the remnant notochord cells of Shh-mutants or a molecular profiling approach might help to illuminate the hierarchy of these genes in sheath formation. On the other hand, a partial contribution of sclerotomal cells to sheath development cannot be completely ruled out at present.
As for the function(s) of the sheath, a logical postulation would be that it serves to restrict and contain the notochord cells in the midline, in a continuous form. In fact, in Smo mutants, notochord cells were seen to be dispersed in the VB region in the absence of the sheath [44]. A similar observation was made in the Sox5-/-Sox6 -/- mutants where the notochord cells were ectopically located [16].
Nucleus Pulposus formation
Around E13.5, the notochord begins to segregate along the anterior-posterior (A/P) axis, showing early signs of expansion into the IVD anlagen. It regresses in the VB regions and expands into the IVD anlagen to form the nucleus pulposus. A distinct nucleus pulposus structure is apparent by E15.5 (Figure 1), filled with notochordal cells and large vacuolated cells [16,20,25]. Whether the notochord cells undergo apoptosis in the VB region and proliferation in IVD or are simply pushed into the IVD region are debatable theories and are discussed in the later part of this review.
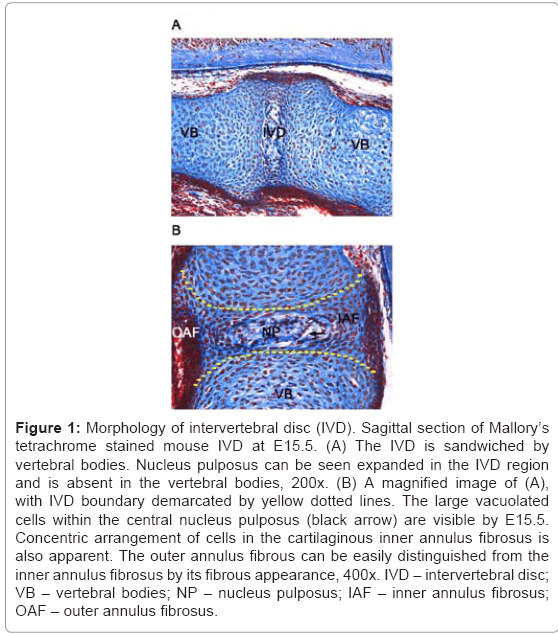
Figure 1: Morphology of intervertebral disc (IVD). Sagittal section of Mallory’s tetrachrome stained mouse IVD at E15.5. (A) The IVD is sandwiched by vertebral bodies. Nucleus pulposus can be seen expanded in the IVD region and is absent in the vertebral bodies, 200x. (B) A magnified image of (A), with IVD boundary demarcated by yellow dotted lines. The large vacuolated cells within the central nucleus pulposus (black arrow) are visible by E15.5. Concentric arrangement of cells in the cartilaginous inner annulus fibrosus is also apparent. The outer annulus fibrous can be easily distinguished from the inner annulus fibrosus by its fibrous appearance, 400x. IVD – intervertebral disc; VB – vertebral bodies; NP – nucleus pulposus; IAF – inner annulus fibrosus; OAF – outer annulus fibrosus.
Lineage-tracing experiments by Choi et al. using Shhcre and ShhcreERT2 mouse lines and a very recent publication by McCann et al. using a Noto-cre mouse line (which is specific to notochord cells) have put to rest the long-standing debate on the origin of the large vacuolated cells of the nucleus pulposus. These authors have shown conclusively that the notochord cells give rise to the nucleus pulposus. Even the small chondrocyte-like cells seen to populate the adult nucleus pulposus in degenerated discs, are derived from the notochord cells as shown by McCann et al. [20,25]. The implication of this finding is enormous, as a decline in the notochord cell population and an increase in chondrocyte-like cells in the nucleus pulposus has been attributed to the development of disc degeneration [2,6,54]. Whether these chondrocyte-like cells are an outcome of aberrantly transformed notochord cells or derived from the large vacuolated nucleus pulposus cells is unknown. The identification of crucial factors involved in the normal differentiation or anomalous transformation of notochord cells as seen in intraosseous benign tumors [25] would greatly assist in the development of cell therapy for DD as well as comprehend its etiology.
As the notochord is formed, the surrounding paraxial mesoderm is segmented into somites and patterned to give rise to the sclerotome or dermomyotome depending on the signals received. These sclerotomal cells then migrate and condense around the notochord, giving rise to metameric condensed and less condensed segments. The condensed regions represent the IVD anlagen, while the less condensed portions give rise to the future VBs. These sclerotomal cells thus give rise to the inner hyaline-cartilaginous and outer fibrous annulus fibrosus of the embryonic IVD [55]. In mutants where the sclerotomal specification, proliferation, viability or differentiation is disrupted, the IVD is either reduced in size or completely absent.
Sclerotome specification
Shh signal emanating from the notochord and the floor plate of the neural tube directs the ventral somites to a sclerotomal fate. It acts by inducing the expression of Pax1 and Pax9 and Mesenchyme forkhead 1 (Mfh1) which mediate its proliferative function [56-59]. Additionally, this requires the maintenance of a BMP-reduced zone by the BMP antagonists Noggin (Nog) and Gremlin (Grem1) in the ventral somites [60,61]. Loss of Nog and Grem1 renders the somites unresponsive to Hh signaling which results in the failure of sclerotome specification. Likewise, mutants where Shh signaling components like Smo [62], Gli2 and Gli3 are impaired do not activate the expression of sclerotome markers [63]. The fact that Nog also induces Pax1 expression in the absence of Hh signaling, and that some Pax1 expression is still detected in Shh-/- embryos, indicates the presence of two, potentially parallel, pathways in sclerotome specification [56,60,64].
Sclerotome maintenance
Maintenance of a certain critical number of sclerotomal cells is crucial for condensation to take place [65]. Pax1, Pax9 and Mfh1 are TFs well known for their importance in sclerotome cell proliferation but dispensable for sclerotome formation. In Pax1-/-Pax9-/- mutants, a marked reduction in cell proliferation rates and an increase in apoptosis was observed, which resulted in a complete absence of VB and IVD. It is postulated that Pax1 and Pax9 may be essential to maintain a crucial number of sclerotome cells permissive for condensation to initiate, upon which endochondral ossification occurs [48]. Surprisingly, in Pax1-/-Mfh1-/- mutants, only proliferation rates were affected but not apoptosis. Thus, Pax1 and Mfh1 synergistically regulate the mitotic activity of the sclerotome cells [59].
Sclerotome differentiation
The annulus fibrosus and VB are derived from a specified pool of sclerotomal cells. While extensive studies have been carried out to delineate the pathway leading the sclerotome cells to VB fate, there is paucity in the knowledge on the mechanisms underlying annulus fibrosus fate-determination.
To a VB fate
Mesenchyme homeobox 1 (Meox1) and Mesenchyme homeobox 2 (Meox2) are needed for the differentiation of sclerotome cells but not their specification. While the sclerotome specification marker Mfh1 was still expressed in the Meox1-/-Meox2-/-mutants, expression of Pax1 and Pax9 was lost in the sclerotome [66]. After specification, Nkx3.2 is required for the proper differentiation of prechondroblast into chondrocytes in the VB. Known markers for chondrogenic differentiation - Sox9, Col2a1, and Fibroblast growth factor receptor 3 (Fgfr3), were down-regulated in the Nkx3.2-/- mutant vertebral anlagen [15]. Interestingly, both Meox1 and Pax1 have been shown to bind and transactivate the Nkx3.2 promoter, indicating their ability to directly activate Nkx3.2. The loss of Nkx3.2 and Pax1 expression in Meox1-/- Meox2-/- embryos further indicate that Meox genes are upstream in the genetic cascade of sclerotome differentiation [66-68].
To an annulus fibrosus fate
Mfh1 and TGF-β signaling are known to play vital roles in annulus fibrosus fate determination. Disruption of Mfh1 or TGF-β signaling led to an abnormal or reduced annulus fibrosus [18,69]. Additionally, gene expression profiling of IVD tissue from a conditional Tgfbr2 knock-out specific to Col2a1-expressing cells showed that the genetic profile clustered more closely with the wild-type VB than the wildtype IVD. These results corroborated the histological observations made in a prior study by the same group on the Tgfbr2 mutants. Also, Fibromodulin (Fmod), an IVD marker, was proven to be a downstream target of TGF-β signaling. The authors thus proposed a potential role of TGF-β signaling in annulus fibrosus differentiation and prevention of chondrocyte differentiation in the IVD region [16,59,69-71].
The expression of Pax1 in the inner and outer annulus fibrosus at E15.5, and the complete absence of IVD structure in Pax1-/-Pax9-/-, suggests their potential role in annulus fibrosus formation as well. It is noteworthy that such distinct features of inner and outer annulus fibrosus are visible only at embryonic stages and that in the adult IVDs the annulus fibrosus is uniformly fibrous [71]. The reasons for such distinction at an embryonic stage and how it is resolved in the adult stage remains to be identified.
The dispute on the origin of cells within the nucleus pulposus (large-vacuolated and small chondrocyte-like cells) may have been resolved with the studies by Choi et al. and McCann et al. [20,25]. Nonetheless, the morphogenesis of the nucleus pulposus structure itself is yet to be elucidated. Two schools of thoughts exist in the field: 1) regional apoptosis and proliferation of notochord cells lead to notochord removal in VB and expansion in IVD region respectively; 2) pressure exerted by the surrounding developing VB on the notochord is responsible for pushing the notochord cells into the IVD region.
In the first scenario, differential proliferation of notochord cells in the IVD space and a concurrent apoptosis in the VB region is considered to contribute to the regional expansion and regression of the notochord. Nevertheless, Azodi et al. failed to observe such a phenomenon in their analysis of embryonic wild-type notochord. Then again, their analysis was restricted to mainly one developmental stage (E13.5) and a region-specific statistical analysis was not performed [72]. Therefore, proliferation and apoptosis assays with statistical evaluation need to be performed in a range of developmental timepoints in both the wild-type and nucleus pulposus-defective mutants, in order to irrefutably disprove this hypothesis.
The premise of the second hypothesis largely relies on the timely coincidence of two events: the notochord expansion and the hypertrophy of the VB, both of which occur at E14.5. It is believed that the accumulation of extracellular matrix rich in proteoglycans in VBs undergoing hypertrophy enables water absorption / retention. This osmotic swelling coupled with a resistance by collagen fibrils confers a pressure on the notochord that runs through the middle of the vertebral bodies. The pressure thus pushes the notochord cells into adjacent IVDs, which results in the characteristic expanded form of the nucleus pulposus [72]. Studies on mutants with abnormal VBs such as Col2a1-/-, Pax1-/-Pax9-/-, and Shh-/- mice also add validity to this hypothesis, since in all of these mutants the notochord fails to expand into the IVD region despite its presence. Furthermore, in conditional Smo and Sox5+/-Sox6-/- knock-out mutants where the VBs are still present, the notochord cells become dispersed in the VB region. This pressure-induced dislocation of notochord cells seems a logical explanation [16,44,48,72].
A third possibility that cannot be completely ruled out is a reciprocal signaling by the annulus fibrosus to the notochord to instruct it to expand into the IVD region. As observed in the conditional Tgfbr2 knock-out mutants, even though the nucleus pulposus is successfully dismantled in the VB, it is not completely expanded into the IVD zone. These mice do not show an overt defect in the VB or their ability to hypertrophy. Hence, the mechanical pressure theory does not account for this defective expansion. Since the inner annulus fibrosus fails to form in these Tgfbr2 mutants, it might be responsible for providing instructive signals to the notochord. Else, TGF-β signaling itself might play a partial role in regulating notochord expansion [69].
The IVD development is an intricate and a fundamental aspect of axial skeletogenesis in vertebrates. In this review, we have discussed the known key mechanisms involved in the formation of various components of the embryonic IVD. Evidently, numerous genes orchestrate IVD development and many more remain to be identified. How all of these genes are networked together to regulate the appropriate cells numbers and control their differentiation in a timely manner are the ultimate questions that deserve attention. In addition, the different hypotheses that have been examined in this review reflect the areas in IVD morphogenesis that ought to be investigated in the future.
More importantly, with the advent of state-of-the-art highthroughput technologies such as RNA-sequencing (RNA-Seq), chromatin immuno-precipitation sequencing (ChIP-Seq) and chromatin interaction analysis with paired-end tag sequencing (ChIAPET), it is apparent that innumerable factors (miRNAs, non-coding RNAs, and cis-regulatory elements), other than genes, regulate the molecular mechanisms of developmental processes. For that reason, it is imperative that we do not restrict ourselves to identifying genes, but broaden our horizon by probing deeper to discover other regulatory molecules that are key players in IVD development. Not only would that equip us with more ways than one to manipulate the genome, we would be able to fine-tune the cell therapy process for DD as well as for other diseases.
We are grateful to Petra Kraus and the anonymous referees for their valuable suggestions on this manuscript.