Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Thesis - (2023)Volume 12, Issue 2
A bioinformatic strategy was proposed for linking ligand Protein Data Bank (PDB) structural fragments to ChEMBL IC50 bioactivity of Protein Kinases (PKs). A bootstrap procedure, based on exhaustive enumeration, was used to assemble, and statistically evaluate, sets of fragments that were enriched for ligands that target PKs in separate branches of the kinome tree. Results found that probes comprised of six fragments return 84% correct predictions for branch-selective PKs ligands. Self-organizing maps were used to cluster the enriched six-fragment probes and, separately, the ChEMBL IC50 data, to identify branch-selective fragments and branch-chemoselective ligands. A contingency table, based on the co-occurrence of branch-chemoselective ligands possessing branch-selective fragments, used for Fisher’s exact tests of independence, found an average recovery of 44% for branch-chemoselective ligands. Seven percent (7%) of these cases represent exact structural matches to PDB ligands, inclusive of eight Food and Drug Administration (FDA) approved oncology compounds. Binding site analysis of enriched branch-selective fragments for these FDA ligands found roles for key hydrophobic and non-hydrophobic interactions. Global extension of these results found a subset of 402 branch-chemoselective ligands with enriched branch-selective fragments, but without crystallographic data, as candidates for selectively targeting PKs. These results extend the use of fragment-selective mining of chemical libraries aimed at discovering ligands that target PKs in separate kinome branches.
Bioinformatics; Self-organizing maps clustering; Fisher’s exact testing; Ligands
Fragment-based Drug Discovery (FBDD) is a powerful tool for discovering leads into small molecule therapies for many human diseases [1,2]. The successful application of FBDD is evident from the nearly three thousand publications currently listed in PubMed Central. FBDD begins with a selection of smaller than ligand substructures that are linked together, chemically or in-silico, to efficiently propose drug leads; leveraging the reduction of chemical space due to fragment-based (versus atom-based) explorations. A significant factor in this success has been the utilization of ligand-target interaction architecture, especially when seeking selectivity [3-5]. Advances in synthetic chemistry have also contributed to FBDD’s successes by generating large fragment libraries for testing [6]. FBDD offers a broad range of strategies, including, but not exclusive to, knowledge-driven focused optimizations of potent fragments and fragment-selective surveys of large chemical libraries or bioactivity databases [7,8].
Protein kinases (PKs) are exceeded only by G-protein coupled receptors as highly desirable therapeutic targets [9,10]. PKs have vital roles in cellular signaling, making small-molecule mediated interference in signal transduction a highly sought-after goal [11-13]. Ligands that target PKs achieve potency by having strong binding interactions, typically through hydrophobic and hydrogen bonding [14]. Finding potent inhibition begins the process of achieving selectivity, often addressed using apparently limitless medicinal chemistry modifications. Crystallographic PKs complexed with small molecule ligands provide atomic indicators for potency and selectivity. Previous studies provide details of PKs-ligand architecture and others make this information universally available [15,16].
Diverse approaches are reported within the body of FBDD literature for exploiting PKs ligand binding sites. Notable is the KinFragLib effort of Volkamer, et al. [17], where chemically synthesizable fragments are used as probes into different regions of the PK’s ligand binding cleft. A slightly different design can be found in the work of Lunney, et al. [18] where the Pfizer internal crystal database was mined to compile fragments that bind to PKs binding pocket. Computational explorations of core structures as templates for molecular design appear in Dimova, et al. [19] with supporting commentary in Hu, et al. [20]. Many of these FBDD designs are aimed at producing large numbers of potentially testable fragments (>7k for KinFragLib and uncountable for synthetic 3D fragment libraries).
Although the successes of FBDD are widely published, seeking improvements that advance the current efforts remains an active research goal [21,22]. One area of improvement focuses on assessing the performance FBDD for identifying biologically active ligands within publicly accessible databases. Strengthening this structure-activity interface will further enhance the utility of FBDD, by providing novel search strategies for identifying ligands that target kinome proteins. With these goals in mind, this study proposes a joint analysis of kinome ligand fragments and their association with biological activity. A two-step design will be used to apply a bootstrap procedure that assembles and statistically evaluates sets of fragments that are enriched within ligands that target PKs in separate branches of the kinome tree and also assess whether these kinome branch-selective fragments can be used to mine existing databases for ligands with kinome branch-specific chemoselectivity.
The components of this analysis will first, explore ligand fragments, derived from existing crystallographic complexes to determine statistically validated fragment subsets that are enriched for ligands targeting PKs of each kinome branch [23]. Second, enriched ligand fragments are used for fragment-selective surveys of compounds within public databases aimed at identifying and statistically evaluating their co-occurrence with kinome branch-chemoselectivity. The overarching purpose of this design is to link ligand structural features, via their fragments, to PK ligand chemoselectivity.
Study population
PKs constitute one of the largest protein families with over 500 members encoded in the human genome [24,25]. Kinome-targeted inhibitor discovery seeks to find ligands that can bind to specific kinases, notwithstanding the fact that all catalytic kinase domains share a common folding motif [26]. Two publicly available data sources are used such as the Kinase Ligand Interaction Fingerprints and Structures (KLIFS) database and the Chemistry European Molecular Biology Laboratory database (ChEMBL). The first database includes the available crystallographic structures of PKs and their co-crystallized ligands. Eukaryotic PKs have been classified based on their sequence similarity into seven main branches such as protein kinase AGC (AGC), Calmodulin/calcium regulated Kinases (CAMK), Casein Kinase1 (CK1), CMGC, STE, Tyrosine Kinase (TK) and Tyrosine Kinase-Like (TKL). Sequence variations across kinome branches, combined with advances in protein and cell-level experimental techniques and an increase in structure-based knowledge of PKs, finds binding site variations between PKs in kinome branches [27,28]. Zhao, et al. [29] have proposed target-selective binding architectures for these major kinome branches. Linden, et al. [45] has also comprehensively examined the inhibitor binding site architecture. Across-branch profiling has also been used to repurpose Imatinib as an adjuvant treatment for Gastrointestinal Stromal Tumors (GIST). The KLIFS database has made the PKs crystallographic information freely available, catalogued across the major kinome branches. Downloading the KLIFS data yields 3832 PKs ligands for the seven major kinome branches. The major branches with PKs for arms having a unique KLIFS ligand are highlighted according to branch color. The Simplified Molecular Input Line Entry Specification (SMILES) representation of these ligands serves as input for the derivation of fragments. All SMILES have been converted to a canonical format using OpenBabel [30].
Second, the ChEMBL database (www/chembl.org) provides bioactivity screening results for small-molecule inhibitors of kinome PKs [31]. The available dataset (ChEMBL_28) for kinome ligands and their targets consists of >500,000 IC50 (uM) measures across >500 proteins. Filtering these records to eliminate ChEMBL ligands with few bioactivity measures (<5) and records with an average IC50 less than 5.0 and a standard deviation below 0.5, yields 22,635 ChEMBL ligands with IC50 measures across a total of 461 PKs. The threshold-based pruning used here is arbitrary but attempts to capture higher IC50 ligands while excluding records with few measures. This data will be used for associating sets of branch-selective fragments to branch-selective chemosensitivity.
Ligand fragmentation
The Rcdk package in the R programming language is used for ligand fragmentation. Typically, Murko fragments are considered in many Structure Activity Relationships (SAR) studies [32,33]. The inventors of this concept define a ring system as one or more rings sharing an edge, and a framework as the union of rings plus linker atoms connecting them [34]. Rcdk generates fragments for rings and linker atoms, requiring, here, a minimum of three heavy atoms. Due to the importance of connecting atoms in ring systems for binding of ligands to their protein kinase targets, the fragments used for analysis consist of rings and their connecting atoms (referred to as linkage atoms) [35]. Throughout this analysis these sub-structures will be referred to as kinome fragments, or more simply, fragments. There are 6347 Rcdk-derived fragments for the 3832 KLIFS ligands. Figure 1 displays an example set of parent ligands and their Rcdk-derived fragments. The parent ligands displayed in this example are all associated with PKs in the AGC kinome branch. These ligands target PKAc and have the Protein Data Bank (PDB) assigned names of 4uja, 4uj2, 4uj9 and 4ujb, with NVX, NVV, S3N and BBQ as their ligand names. The six fragments associated with these ligands are displayed after the parent ligands. Inspection reveals that inclusion of linker connected fragments results in shared substructures. The analysis proposed here will treat each fragment as a separate chemical entity (Figure 1).

Figure 1: 2D renderings of three kinome ligands that share six RCDKderived fragments. The parent ligands are all in the AGC kinome branch. Labels for parent ligands include the kinome branch:PDB target: PDB name:chain:ligand name. These ligands target PKAc and have the PDB targets with assigned names of 4uja, 4uj2, 4uj9 and 4ujb, and NVX, NVV, S3N and BBQ as their ligand names. The fragments are assigned an arbitrary index (1 through 6347). In this example, fragment indices 356, 358, 357, 26, 1 and 9 are ordered by molecular weight. These indices and their SMILES appear below each 2D rendering.
Fragment survey
The 15 most frequent KLIFS fragments are displayed in Figure 2, where possible International Union of Pure and Applied Chemistry (IUPAC) names are assigned. A sample of fragment canonical SMILES, chemical names, and IUPAC names are mentioned. As expected, phenyl (fragment 1) is the most frequent (n>2k). In decreasing order of frequency, fragment 44 is pyridine, fragment 226 is pyrimidine, a fragment 32 is 7H-purine and fragment 31 is tetrahydrofuran. Fragment 13 is piperazine, fragment 33 is a 9-(oxolan-2-yl) purine pyridine and fragment 38 is piperidine. Fragments 182 and 125 are different tautomers of 1H-pyrazole. Fragment 257 is N-phenylpyrimidin-2-amine, fragment 293 is cyclopropane, fragment 221 is 1,3-thiazole, fragment 299 is morpholine and fragment 412 is cyclohexane. When possible, IUPAC fragment names will be referenced, however, fragment indices (1 through 6347) with be used throughout the manuscript (Figure 2).

Figure 2: Summary of the 15 most frequent fragments in the KLIFS kinome dataset. Top panel displays the histogram count. Bottom panel displays 2D renderings of these fragments and their associated indices. Fragments with IUPAC names appear in the text.
Analysis of fragments utilize a Boolean data matrix consisting of 3832 rows, labelled according to the KLIFS ligand description (branch:target name:chain:ligand name), and 6347 columns (labeled by fragment indices). The cells in each row are true if the ligand contains a fragment and false if it does not. The mean number of fragments per ligand is 5.95 ± 3.73, with a median of 6 fragments per ligand. The determination of fragment enrichment is based on whether the sets of ligands within each kinome branch (referred to as hits) have fragments that are statistically different from fragments in the rest of the kinome branches (i.e., non-hits). Branch-specific hits are statistically assessed for enrichment using a Fisher’s exact test of independence.
Fragment enumeration
The Boolean data matrix, consisting of 3832 rows (number of KLIFS ligands, each identified by its kinome branch) by 6347 columns is used for analysis. Although the median number of fragments per ligand is ~ 6, variations exist, inclusive of fragment counts per ligand a high as fifteen and as low as one. The effect of these variations on designing a systematic analysis of fragments across branches of the kinome tree is illustrated. The upper panel represents a case where three fragments appear in eight KLIFS ligands that target three kinome branches (AGC, CK1 and CMGC). The lower panel represents a case where many fragments (n=12) appear in only six KLIFS ligands, all in the TKL branch. In this latter case, only subsets of these twelve fragments appear within the six TKL ligands. These extreme cases highlight the need to develop a standardized procedure for fragment analysis (Figures 3A-3C).

Figure 3: Represents 2D renderings of parent and fragment structures. (A): Displays the case of few (n=3) fragments appearing in eight ligands spanning three kinome branches.

Figure 3B: Fragments are labelled by their index (1-6437) and molecular weight. Parent ligands are labelled by their index (1-3832 PKs), kinome branch, PKs name, PDB name for PKs, PKs chain and PDB name for ligand.

Figure 3C: Lower panel represents the case of twelve fragments associated with six ligands in the AGC kinome branch.
The proposed strategy is to enumerate all possible fragment combinations per ligand, centered on the population median of six; and extended above and below this median, across a range of 4 to 8 fragments. For future reference, the enumeration results will be referred to as 4-mers, 5-mers, 6-mers, 7-mers and 8-mers. Enumeration of each -mer is completed as an R-script using nested loops. Developing the analysis around these -mers serves two purposes. First, the identification of potential PKs based on the existence of fragment composition of their ligand is generalized to take into consideration the diverse numbers of fragments per ligand. For example, a kinome ligand with twelve fragments could be used to identify candidate ligands in a screening library that possess these twelve fragments. While a ligand with all twelve fragments may exist, absence of a hit would yield no information. Systematic exploration of ligand candidates with fewer fragments can yield screening hits which could be studied further. Second, analysis of -mers can be used to apply statistical tests (e.g., Fisher’s exact test of independence) for each enumeration to identify sets of fragments that are enriched for a specific kinome branch. Therefore, the starting Boolean matrix serves as input for enumeration of all -mers and subsequent statistical testing for their enrichment within each kinome branch.
Fragment enrichment
The process begins with the enumerated fragments (i.e., -mers) derived from KLIFS. Typically, fragments associated with bioactivity are used to conduct SARs. Here, bioactivity is replaced by kinome branch and hits are represented by all rows in this matrix that are in a kinome branch (either AGC, CK1, CAMK, CMGC, STE, TK or TKL). The Boolean matrix, comprised of true and false fragment assignments, is used to test whether sets of fragments can be statistically associated with hits in a kinome branch. In other words, is a ligand’s status of having or lacking a -mer independent of its status as a hit (i.e., being in a specific kinome branch). A Fisher’s exact test of independence (McDonald, 2014, Handbook of Biological Statistics), applied to each set of enumerated fragments (e.g. -mers), yields a p-value for assessing whether fragments associated with hit ligands are enriched when compared to their appearance in non-hit ligands. Using the example of 6-mers, enumeration of KLIFS ligands sharing six fragments (i.e., 6-mers) is obtained for the 3832 ligands. These results are segregated into 6-mers from ligands within (hits) and excluded (non-hits) from a specific kinome branch, to generate a contingency table for Fisher’s exact testing. Statistically significant cases determine enriched sets of -mers within a kinome branch. Quantile-quantile plots (QQ) plot [36]. The vector of p-values generated for all kinome branches is strongly different from random.
ROC analysis of enriched -mers
Enriched 6-mers can be used to generate ROC curves for determining whether their Area under Curve (AUC) values is significantly different from random [37]. Testing is based on tabulating the enriched fragments within the enumerated results for all ligands containing a specific 6-mer and dividing these ligands into those included and excluded from a kinome branch. ROC generates a curve for the false positive fraction (cases where an enriched -mer occurs for a ligand excluded from a kinome branch) over a range of zero to one. The area under each ROC is used to determine whether the enrichment results are different from random.
Data clustering of fragments and ChEMBL IC50 data
Numerous statistical tools are now available for clustering data [38]. Relying on our prior analysis, the results presented here use Self-Organizing-Maps (SOMs) [39-41]. In general, each SOM node defines a codebook vector representing the average response for members clustered to that SOM node. SOM codebook vectors serve as a basis for comparisons to other SOM nodes and for statistical testing of within kinome branch preferences. Two data sources will be clustered; the first is for fragments with significant Fisher’s exact enrichment scores and the second is for the ChEMBL IC50 data. For purposes of nomenclature, codebook vectors of SOMs for each dataset will be identified using subscripted prefixes; fragSOM and ChEMBLSOM, respectively. fragSOM patterns are used to cluster groups of -mers enriched within a kinome branch, while ChEMBLSOM patterns will be used to cluster compounds with enhanced chemosensitivity for PKs within a kinome branch. While each SOM node represents a cluster of input vectors, these nodes can be optimally grouped into meta-clades, based on similarity. Statistical methods for deriving meta-clades are based on identification of the optimal number of clades from SOM codebook vectors. State-of-the art procedures for finding the optimal number of meta-clades include the elbow, silhouette and gap statistic methods [42].
Identification of ChEMBLSOM nodes with enhanced chemosensitivity
Each SOM codebook vector is divided according to IC50 values within (hit) and excluded (non-hit) from PKs in a kinome branch. A Student’s t-test is used to identify IC50’s of relatively higher chemosensitivity for hit versus non-hit PK IC50 values. Student’s p-values less than or equal to 0.05 were further assessed for statistical significance by Bootstrap resampling comprised of 1000 Student’s t-tests for each SOM node, with random shuffling of each SOM codebook [43,44].
Associating fragments to chemosensitivity
ChEMBLSOM provides the framework for linking kinome branch-specific chemosensitivity, as defined above, to compounds having kinome branch-specific 6-mers. To complete this linkage, the >22k ligand’s fragments are selected that have enriched branch-specific 6-mers. The location of each of these compounds on ChEMBLSOM will be referred to as its projection, which is based on the best matches of a compound’s input data vector (a vector of IC50 measures for 461 PKs). The best projection would apply for perfect input data. The extent to which the IC50 values are less than perfect is difficult to determine, however additional measurements (costly) may aid in estimating data variation. As an alternative, noise can be randomly added to each element of a data vector. For this analysis, noise is obtained from the population distribution of standard deviations for all >22k IC50 vectors. Re-projecting data vectors with 1% error from the population distribution extends the best projected SOM node by an average of ± one SOM nodes (e.g., 2/1056=0.0019).
Flowchart
Figure 4 displays a flow chart summarizing the methods’ vignettes. To review, the left arm of the flow chart uses KLIFS data as input for deriving fragments, enumeration for generating -mers, assessment of statistically enriched branch-selective -mers, clustering (fragSOM) and meta-clade assignments. The right arm collects the ChEMBL IC50 data, identifies the fragments of their ligands, clustering (ChEMBLSOM), meta-clade assignment and identification of kinome branch-selective chemoselective nodes. The two arms merge by associating the enriched branch-selective fragments (6-mers) with branch-selective chemosensitivity using Fisher’s exact testing for significance (Figure 4).

Figure 4: Flow chart summarizing the methods’ vignettes.
Fragment survey
The highest ligand counts are for kinome branches TK (n=1301) and CMGC (n=1231) and mirror the high interest for finding ligands that target many oncology PKs in these branches. An average of 72% of the fragments is unique to each kinome branch (range: 81% for TK to 41% for CK1). The relatively high fraction of unique fragments supports their consideration, separately, or in sets, for mining ligands that might target separate kinome branches. Additional support for kinome branch-specific fragments appears in Figure 5, where histograms display counts for the 10 most frequent fragments in the KLIFS dataset, separated according to the seven kinome branches. These results reveal that fragments are not distributed uniformly, with some fragments being present or absent, depending on the kinome branch. Noteworthy in this example is the appearance of fragment combinations 17, 314 and 487, 488, 59, 490 only in the CAMK and CK1 branches, respectively (Figure 5).

Figure 5: Histogram of most frequent fragment counts for all kinome
branches. The top ten most frequent fragments per kinome branch
are selected (yielding 22 unique fragments). Histogram bars display
the fragment counts across all kinome branches. The legend indicates
branch colors. Histogram bar for fragment 1 (benzene) has been scaled
by a factor of four (Ntot for benzene=2283). 
Summary of enriched -mers
The enrichment results for all -mers across the seven kinome branches are mentioned. In summary, Fisher’s exact filtering retains, on average, ~ 10% of the starting ligands for each kinome branch, ranging from a high of 15.4% for 4-mers and a low of 2.6% for 8-mers. Speculations regarding this high attrition include the fact that many KLIFS ligands have diverse kinome targets, thereby making statistical separations between kinome branches less likely, based on fragment composition. The boxplots in the upper left panel of Figure 6 are used to summarize enriched -mers that correctly identify branch-specific PKs. The PKs for all enriched -mers are collected and stratified according to kinome branch. Each boxplot represents the fraction of correct branch assignments (e.g., the number of correctly assigned PKs divided by the total number of unique PKs for all -mers). These results find that the fraction of correct assignments has the lowest average value (0.46) when using 4-mers and the highest average value (0.84) when using 6-mers. Qualitatively, the use of 5-mers and above yields a fraction of correct cases above 0.7. Superimposed on these boxplots is the fraction of starting ligands retained for each -mer after Fisher’s exact enrichment filtering. This diagonal line represents the attrition discussed above. To review, 4-mers have the largest number of ligands and the poorest success rate, while 6-mers yield the highest success rate (84%) with the numbers of ligands intermediate to the complete range (Figure 6).

Figure 6: Upper left panel displays a composite boxplot summarizing the mean and standard deviation for the fraction of kinome PKs (y-axis) for
ligands are identified as having enriched -mers. The diagonal solid line (square symbols) indicates the relative fraction of starting ligands that survive
Fisher’s exact testing. The five additional barplots represent the distribution of ligand counts for 4-mers through 8-mers. For example, the 7-mer
barplot in the upper right corner finds that all enriched fragments are associated with the AGC branch, whereas enriched fragments associated
within the TK branch also include PKs in the TKL and CK1 branches. 
Also displayed in Figure 6 are five histograms (one for each -mer) where each histogram bar is color-coded according to the fraction of PKs associated with enriched branch-selective -mers. For example, the leftmost red bar in the 7-mer histogram upper right panel in Figure 6 represents the results for the AGC branch. Here enriched 7-mers for the AGC branch find only PKs within the AGC kinome. The remaining histograms for 7-mers consist of mixtures of PKs from other kinome branches (i.e., CAMK (2nd bar) has most of its PKs in the CAMK branch (yellow), with lower numbers of PKs in the STE (blue) and TKL (magenta) branches. In general, the majority fraction of each histogram bar is also associated with each kinome branch (citing the rightmost TKL histogram for 7-mers, where the largest contribution is from PKs in the TKL kinome (magenta)). This persists for all -mers; with qualitative differences. For example, 4-mers have the highest contributions in each bar from PKs in many kinome branches, with no less than 5 branches contributing to each bar in the histogram. In contrast, 6-mers have the fewest number of histogram bars with contributions from other kinome branches. For example, the 6-mer histogram bars for AGC, CAMK, CK1, STE and TKL are dominated by within kinome branch cases, with CMGC and TK having small contributions from PKs in other branches. Qualitatively these results indicate that analyses focused on 6-mers offers the highest potential for assigning PKs to their correct kinome branch.
A further check of PKs ligands identified from the 6-mer enrichment results, finds many that are designated as important therapeutic targets. The number of PKs associated with ligands containing enriched 6-mers is displayed as histograms. Noteworthy are the 35 PKs for the tk kinome branch: with EphA2, ABL1, EGFR and ALK as most frequently occurring PKs. Consequently, while attrition due to filtering for statistically significant enrichment eliminates most of the PKs in the KLIFS database, those that remain include many important therapeutic targets.
ROC analysis
The ROC results for six of the seven kinome branches are significant; with AGC flagged as degenerate due to the absence of false positives in the enriched set. The ROC curves for each kinome branch are displayed. The most significant result is for TKL with a p-value of 9.52e-23 and a fitted AUC of 0.903, while the least significant case is for CK1, with a p-value of 0.0389 and a fitted AUC of 0.621. While the utility of ROC measures of accuracy, sensitivity and specificity remain under debate, the values listed are reasonable, with, for example, accuracy values ranging from 34.7 (CMGC) to 73.9 (TK) [45]. Collectively, these results support the existence of 6-mers specific for each kinome branch.
Data clustering of fragments
Figure 7 displays fragSOM for the 6-mer data. There are 8002 6-mers with significant Fisher’s exact enrichment scores. There are 304 unique fragments within this set of 6-mers. The input matrix for SOM clustering consists of 8002 rose by 304 columns, where the fragments within each 6-mer are assigned the value of one and zero otherwise. Drawing from the analogy of clustering molecular fingerprints, prior SOM analysis has proved an effective clustering tool [46]. The top left panel displays the 35 × 20 fragSOM based on these fragments. The colorbar at the right of fragSOM identifies strongly similar codebook vectors as dark copper, while regions with distinct codebook vectors appear as light copper. The 700 codebook vectors, organized as a heatmap (R utility gplots: heatmap 2, Euclidean, Wards.D2), appear in the bottom image. Clustering of fragments is evident as colored blocks travelling up from the lower left of the heatmap, curving upwards towards the upper right. Rather than analyze each fragSOM node (e.g., hundreds), the codebook vectors are organized into meta-clades. Using the gap statistic in the R programming language, 19 optimal meta-clades are found for the 700 SOM codebook row vectors. Meta-clades based on SOM codebook rows will be referred to using the capital letter ‘C’ followed by the meta-clade number (i.e., C1 through C19). The colorbar at the left of the heatmap identifies C1-C19 optimal meta-clades spectrally from blue to red. The boundaries for these 19 meta-clades appear as red lines in the SOM image (upper left) and are mapped onto the SOM (upper right), color-coded from blue to red according to the colorbar at left of the heatmap. Organizing SOM codebook vectors using meta-clades facilitates the analysis of fragments within kinome branches. There are 32 optimal meta-clades based on the 304 SOM codebook column vectors (designated FC1-FC32). Their dendrogram and meta-clade colorbar appears at the top of the fragSOM heatmap. These results find that meta-clades C1-C8, shown in the blue colors in fragSOM at the upper left of Figure 7, are associated with relatively few numbers of fragments (FC1-FC5). fragSOM meta-clades colored in yellow-green-red on fragSOM are associated with column-derived meta-clades (FC6 and above) that consist of diverse fragment types. Upper right panel in Figure 7 transfers the 19 row-based meta-clades to fragSOM, preserving the coloring in the bar to the left of the heat map. These boundaries appear as red lines in fragSOM displayed in the upper left panel (Figure 7).

Figure 7: Upper-left panel displays fragSOM generated from the matrix
of 6-mers (8002) by 304 unique fragments. fragSOM is colored in copper
where regions of light copper indicate diverse fragments, while dark
regions indicate cases of similar fragments. The fragSOM dimensions of
35 × 20 yield 700 codebook vectors, each with 304 fragments. fragSOM
represent clusters of 6-mers with similar fragments. Each fragSOM
node represents the average of the 304 fragments in each cluster. The
lower panel displays the heatmap of codebook vectors. Heatmap colors
define codebook vectors from high (yellow) to low (purple). fragSOM
nodes lacking a fragment appear as white. Dendrograms (Euclidean,
Wards.D2) appear to the left and above the heatmap. Clusgap analysis
(R:cluster::clusgap) of this heatmap finds 19 meta-clades (C1-C19)
for the codebook rows and 32 meta-clades (FC1-FC32) for codebook
columns. The colored bars at the left and above the heat map identify
these meta-clades (spectrally color-coded from blue to red). 
The fragSOM clustering of enriched -mer data organizes this data according to -mer composition. Each fragSOM node’s cluster members include labels for the parent ligand containing each -mer and its kinome branch. Meta-clades are proposed for grouping the clustered data. Therefore, meta-clades utilize codebook vectors, organized into an optimal number of meta-clades, to provide input for analysis. fragSOM analysis for each -mer finds a consistent number of optimal meta-clades (4-mer:23, 5-mer:25, 6-mer:19, 7-mer:19 and 8-mer:22). The fractional contribution of kinome branch members of all -mers is displayed as histograms. Inspection reveals that each histogram has meta-clade bars composed of progressively fewer numbers of mixed kinome branches with increasing -mer count. Inspection also reveals that the results for 7-mers and 8-mers appear to have achieved clustering due to a bias towards relatively greater numbers of members in the TK kinome branch. In contrast, the 5-mer and 6-mer results yield comparable numbers of optimal meta-clades, with 6-mers having the fewest number of optimal meta-clades with mixed kinome branches. These results support the use of 6-mers for further analysis. For comparison to the fragSOM results, clustering of the 6-mer data was also completed using Stochastic Neighbor Embedding (R::Rtsne). To summarize, hierarchical clustering of Rtsne results was unsuccessful at separating kinome branch members based on fragment composition.
Data clustering of ChEMBL IC50 data
The ChEMBL dataset, comprised of 22635 ChEMBL compounds with IC50 measures against 461 PKs, is used to generate ChEMBLSOM. Figure 8 displays ChEMBLSOM (upper left panel), the heatmap of codebook vectors (upper right panel) and the regions associated with the optimal meta-clades (red boundaries and vertical colorbar). SOM tools (based on the ratio of the first 2 principal components of the data matrix) determined a map size of 44 rose by 24 columns. Each of these 1056 SOM nodes identifies clusters of ChEMBL compounds with similar IC50 measures. In this regard, the >22k records analyzed using SOMs results in a 22-fold data reduction. ChEMBLSOM nodes are colored in copper (upper left panel), indicating codebook vectors that are similar (dark copper) and distinct (light copper). Observation finds that the distinctive codebook vectors are mostly located around the perimeter. The upper right panel displays a heatmap (R utility gplots::heatmap.2) for codebook vectors, with the dendrograms at the left and top representing row and column clustering of 1056 codebook vectors and 461 response vectors, respectively. The colored bar adjacent to the leftmost dendrogram identifies, spectrally (blue to red), the 22 optimal meta-clades for the codebook vectors (R::clus_gap, Euclidean, Wards.D2, Hartigan-Wong). The red boundary lines in the upper left panel identify the regions associated with these optimal meta-clades. For comparison, associative clustering (R utility apcluster) of these codebook vectors also finds the optimal number of 22 meta-clades [47,48]. The 22 optimal meta-clades are mapped to ChEMBLSOM displayed in the lower left corner, organized spectrally from blue to red.

Figure 8: Upper left panel displays ChEMBLSOM for the 22635 × 461 input matrix. ChEMBLSOM is colored in copper where regions of light
copper indicate diverse IC50 values, while dark regions indicate cases of similar IC50 measures. Each ChEMBLSOM node represents a cluster of
ChEMBL compounds with similar IC50 values. The upper right panel displays the heatmap for codebook vectors (each vector has 461 members).
Heatmap colorbar represents low to high IC50 values spectrally from purple to orange. R::Clusgap analysis of codebook vectors finds 22 optimal
meta-clades (Euclidean, Wards.D2, Hartigan-Wong), designated as C1-C22. The color bar at the left of the heatmap identifies these 22 meta-clades
spectrally from red to blue. The image in the bottom left maps the optimal meta-clades. The lower right panel displays a histogram for the topmost
15% of significant ChEMBLSOM nodes for the 22 meta-clades. These results support the clustering of codebook vectors into different meta-clades. 
The lower right panel in Figure 8 displays a histogram for the topmost 15% of significant ChEMBLSOM nodes for the 22 meta-clades (C1-C22). Inspection finds meta-clades comprised of a single kinome branch (C3, C15 and C17 for CAMK, TK and TKL, respectively) and mixtures (C6 and C13 are comprised of TK and TKL members, C14 is comprised of AGC and CMGC members). At the level of ChEMBLSOM meta-clades, significant nodes based on branch-selective chemosensitivity are comprised of a majority fraction from only one kinome branch (Figure 8).
Identification of SOM ChEMBL nodes with enhanced chemosensitivity
Figure 9 illustrates the kinome branches AGC and TKL. ChEMBLSOM is depicted as a wireframe of (44 × 24) hexagons, where colored hexagons designate, as the t-statistic, significant ChEMBLSOM nodes. Each panel identifies the statistics for each node. For example, node (1,13) for AGC has a statistical significance of 4.583e-3 when 39 IC50 values for PKs in the AGC branch are tested against the 103 IC50 values not in the AGC branch. The sorted IC50 values appear as adjacent histograms, where the within-kinome-branch IC50’s are depicted in black (Figure 9).

Figure 9: Summary of results for ChEMBLSOM for kinome branches AGC (left panel) and TKL (right panel). ChEMBLSOM appears as a wireframe of hexagons, where only the significant Student’s t-tests are colored as their t-statistic. Adjacent to each ChEMBLSOM are histograms of IC50 activity for nodes (1,23) and (20,13) for AGC and nodes (9,2) and (30,11) for TKL. Colorbars correspond to the Student’s t-static values, (red: high and blue: low).
Associating fragments to chemosensitivity
The intersection of ChEMBLSOM projections with nodes having branch-specific chemosensitivity is used to construct a contingency table for Fisher’s exact testing. These results report the p-values for these tests and their corresponding contingency matrix (true positives, false positives, true negative and false negatives). An example for TKL appears in Figure 10. The upper and lower panels display the TKL results for perfect and noisy projections, respectively. The panel at the left displays the ChEMBLSOM TKL-chemosensitive nodes (n=86), while the middle panels display the perfect (upper; 224 chembl 6-mers, 82 nodes) and noisy (lower; 241 chEMBL 6-mers, 323 nodes) projections for ChEMBL compounds containing TKL-specific 6-mers. The right-most panel identifies the intersecting nodes used as the true positive value in the contingency table (n=17 and 42, respectively). The Fisher’s exact p-values for these cases are 1.398e-4 and 1.642e-4, respectively. Inspection of the ChEMBLSOM projections for noisy data in Figure 10 may appear to increase the likelihood of more true positives. However, by design, the contingency table for Fisher’s exact testing accounts for true positives/negatives and false positive/negatives. As a result, projections, when applying noise to the data, will influence all members of the contingency table. Noteworthy is the comparable Fisher’s exact p-values for perfect and noisy data. Noise-perturbed data does, however, yield greater numbers of true-positive ChEMBL compounds when compared to perfect data (Figure 10).

Figure 10: Summary of results for ChEMBLSOM projections for kinome branch TKL. ChEMBLSOM appears with the 22 optimal meta-clade boundaries shown as solid lines. Left-most panel shows ChEMBLSOM nodes that have significant Student’s t-test values for TKL (n=86). The middle panels in both rows display the projection nodes for ChEMBL compounds containing the 6-mer probes associated with each kinome branch, based on perfect data(n=82) and noisy data (n=323). The right-most panels identify the intersection ChEMBLSOM nodes from the left and middle panels (n=17 and 42, respectively). On average 58% (13143/22635) of the ChEMBL compounds are associated with chemosensitive ChEMBLSOM nodes. Within these compounds, 8% (984/22635) have branch-selective 6-mers. These compounds project to 230 and 683 ChEMBLSOM nodes based on perfect and noisy data, respectively.
Cluster-based association of fragment composition to kinome branch
The following analysis is used to associate sets of fragments (i.e., -mers) to a kinome branch. The enriched 6-mers used for fragSOM clustering represents all unique sets of 6-mers that pass the Fisher’s exact enrichment test. Ligands with more than six fragments included all combinations fragments in the enumerated 6-mers. Such cases will have five core fragments, plus all combinations of fragments above five that exist for a ligand, that also pass the Fisher’s exact test. Consequently, if a ligand has ten fragments, enumeration yields five records composed of the core five fragments, repeated for each of the five additional fragments from its set of ten. The 32 fragment meta-clades (R::clusgap), labelled as FC1-FC32, and their fragment membership (as fragment indices) are listed in the top panel of Figure 11. The colorbar at the right of this panel is replicated (inverted) from above the heatmap in Figure 7. The lower panel in Figure 11 displays the dendrogram for the 6-mer fragments (replicated from Figure 7). Noteworthy is the existence of 15 meta-clades with only one fragment. In contrast, meta-clade FC32 consists of 163 fragments. The distribution of fragment memberships in meta-clades FC1-FC32 will be analyzed later based on the 19 ChEMBLSOM meta-clades (Figure 11).

Figure 11: Top table lists the fragments for each of the 32 meta-clades (labelled FC1-FC32). Color bar at the right corresponds to the 32 metaclades (R:clusgap). Dendrogram at the bottom is based on the fragSOM column vectors. The horizontal and vertical color bars represent the 32 optimal meta-clades. Two-hundred and forty-seven of the 304 (82%) KLIFS 6-mers exist in the ChEMBL fragments. There are 3119 mutual fragments between the KLIFS (3119/6347=0.491) and CHEMBL (3119/23411=0.133).
The top panel in Figure 12 displays the 6-mer histogram for the 19 fragSOM meta-clades (labelled C1-C19) identified (R::clusgap) for fragSOM. The histogram bars are color-coded according to kinome branch PKs membership (discussed in Methods: summary of enriched -mers). These results find thirteen fragSOM meta-clades with memberships associated with only one kinome branch (TK:C1,C2,C3 AGC:C4 TKL:C6,C9,C16 CK1:C7 CMGC:C8,C13 CAMK:C11,C17 STE:C15). In contrast, fragSOM meta-clade C19 consists of a mixture of six kinome branches. Based on these results, fragment compositions exist for PKs within a single kinome branch and shared between kinome branches. The middle panel in Figure 12 lists the meta-clades C1-C19 (column 1) the fragments associated with these fragSOM meta-clades (column 2), the number of 6-mers (column 3) and the kinome branch (column 4). Fragments associated with meta-clades are compiled from the sets of enriched branch-selective 6-mers. The most frequently occurring fragments, ordered from the highest frequency, are 226,1,13,44,38,88 and 459 and are highlighted in yellow. Inspection would indicate that the most frequently occurring fragments do not appear to constitute fingerprints for specific kinome branches. However, sets of fragments (e.g., 6-mers) may represent a more powerful screening tool when compared to fragment frequency.

Figure 12: Left histogram represents the branch membership for the 19-meta-clades (C1-C19) from fragSOM. Right histogram displays the
distribution of the FC1-FC32 fragment meta-clades for each of the 19 fragSOM meta-clades, colored according to FC composition. 
The lower panel in Figure 12 displays a distribution histogram for the 32 fragSOM meta-clades (FC1-FC32) across the 19 fragSOM meta-clades (C1-C19). The colors for FC meta-clades are consistent with the colorbar in Figures 7 and 11. Inspection of this histogram finds a non-uniform distribution of FC meta-clades across the fragSOM meta-clades. For example, FC1-FC8 (depicted as shades of blue) dominate the TK-specific fragSOM meta-clades C1-C3. The middle clusters (C4-C9) include contributions from fragments in FC11-FC19 (shades of yellow-orange). The right-most clusters have contributions from FC20 and above (shades of red). Note that FC32, consisting of 163 enriched fragments, is excluded from this histogram. These results are consistent with the heatmap in Figure 7 and the dendrogram to the left (displaying fragSOM meta-clades C1-C19; also shown in Figure 11) and fragSOM fragment meta-clades FC1-FC32 displayed above the heatmap in Figure 7. Notably, contributions from less frequently appearing fragments are important for ligands that target non-TK kinome branches. These results indicate the importance of less frequently appearing fragments within sets of enriched 6-mers (Figure 12 and Table 1).
| fragSOM meta-clade | Fragment indicies | 6-mers | branch |
|---|---|---|---|
| C1 | 1,13,44,88,226,257,459,2387,2441,2442,2443,2697,2698,2699 | 943 | TK |
| C2 | 1,13,44,88,226,257,459,2387,2441,2442,2443,2697,2698,2699 | 857 | TK |
| C3 | 1,13,17,38,44,88,226,257,374,375,459,1907,2387,2441,2442,2443,2697,2698,2699,4472 | 1208 | TK |
| C4 | 1,38,71,72,73,75,76,77,78,79,81,82 | 925 | AGC |
| C5 | 1,125,226,257,293,522,523,524,525,526 | 210 | CAMK |
| C5 | 1,26,125,226,229,293,509,522,524,525,527,528,529,530 | 217 | STE |
| C6 | 38,182,216,293,2987,3671,3674,3676,3677,3678 | 86 | TKL |
| C7 | 182,226,293,412,1268,1269,1270,1271,1272,1273 | 211 | CK1 |
| C8 | 1,9,13,44,93,226,1662,1988,1993,1995,1998 | 448 | CMGC |
| C9 | 1,13,93,498,499,517,518,519,520,3315,3316,3317,3318 | 212 | TKL |
| C10 | 1,38,498,499,500,501,502,503,504 | 84 | TKL |
| C10 | 1,38,498,499,500,501,502,503,504 | 84 | CAMK |
| C11 | 1,13,38,78,754,755,756,757,758,759 | 211 | CAMK |
| C12 | 1,13,221,226,232,1573,2768,2769,2770,2771 | 210 | TK |
| C12 | 1,221,226,1267,1871,3403,3417,3462,3463 | 84 | TKL |
| C13 | 1,226,521,725,2083,2128,2129,2133,2134,2135 | 211 | CMGC |
| C14 | 1,44,57,59,1310,1311,2001,2002,2003,2004 | 210 | CMGC |
| C14 | 1,59,226,277,1274,1275,1276,1277,1278,1279 | 210 | CK1 |
| C15 | 1,44,893,1699,2652,3213,3214,3215,3216,3217 | 211 | STE |
| C16 | 1,3,44,182,221,299,314,417,423,424,490,498,675,738,877,896,934,941,1118,1285,1379,1452,1453,1454,1482,1592,2558,2562,2564,3137,3144,3869,3870 | 218 | TKL |
| C17 | 1,13,44,93,760,761,762,763,764,765 | 211 | CAMK |
| C18 | 1,13,88,459,539,3116,3511,3512,3513 | 84 | TKL |
| C18 | 1,9,13,88,226,459,507,3116,3118 | 80 | STE |
| C19 | 1,2,3,4,13,38,44,93,125,139,216,293,299,326,468,561,636,637,725,964,965,966,967,1261,1568,1699,2652,3135,3137,3138,3145,3224,3226,3241,3243,3248,3249,3250 | 304 | STE |
| C19 | 1,2,13,38,44,94,125,133,216,221,226,412,513,561,697,759,806,848,1345,15 73,1574,2520,3959,3960,3961,3962,4089,4493,4494,4663,4664,5004,5008,5009 |
198 | TK |
| C19 | 1,2,13,44,93,125,226,396,412,521,787,829,890,982,1029, 3321,3322,3365,3367,3568,3569,3570,3591,3592,3593 |
31 | TKL |
| C19 | 1,13,44,93,125,128,138,226,259,293,498,517,703,1040,1044,1114,1193,1194,1195,1196, 1504, 1662,1818,1829,1834,1988,1993,1995,1998,2025,2027 |
19 | CMGC |
| C19 | 1,2,9,13,26,38,44,93,139,261,299,459,508,517,561,636,637,660,661,662,663,793,794,856,857,858,859,877,934,960,961,962,963,1170,1171 | 13 | CAMK |
| C19 | 1,9,17,26,38,121,129,154,155,156,208,209,226,252,253,254,297,299,300,301,308,309,356,357,358 | 11 | AGC |
Note: CAMK: Calmodulin/calcium regulated Kinases; CK: Casein Kinase; TK: Tyrosine Kinase; TKL: Tyrosine Kinase-Like
Table 1: Table lists the fragment composition for each fragSOM meta-clade.
The following result illustrates of the importance of sets of fragments (i.e., 6-mers) towards identification of ligands that target a specific kinome branch. Thirteen ChEMBL compounds with chemoselective IC50 activity against PKs in the AGC branch are displayed. These PKs ligands contain only three 6-mers: 121, 226, 297, 299, 300, 301 or 38, 121, 226, 297, 308, 309 or 88, 121, 226, 297, 308, 309. While each set of 6-mers contain at least one of the most frequently occurring fragments, the composition of less frequent fragments also plays an important role in targeting to a specific kinome branch.
SOM analysis of chemosensitivity
This result identifies ChEMBLSOM nodes with codebook vectors that have significantly greater IC50 measures for ligands that target PKs in each kinome branch. Recall that each ChEMBLSOM node represents the average IC50 response of the 461 kinome targets. A Student’s t-test is used to conduct comparisons between IC50 measures that are included and excluded from each of the 7 kinome branches. Table 2 lists the results for Fisher’s exact testing for the co-occurrence of ChEMBL compounds with branch-selective 6-mers and branch-selective chemosensitivity (cf. Figure 9 for examples of branch-selective chemosensitivity). P-values range from the best, of 2.08e-5, for CMGC to the worst, of 3.67e-2, for CAMK (using noisy data). These results find a low of only 3 true-positive ChEMBLSOM nodes with projections of 4 ChEMBL compounds for CK1, to a high of 82 chemblSOM nodes with 133 ChEMBL compounds for TK. The former result, as well as the results for kinome branch STE, has the lowest representation in the starting KLIFS dataset and the fewest ChEMBL IC50 data against their PKs. Despite this modest representation, significant Fisher’s exact p-values are found for these as well as the remaining kinome branches. The results for perfect data yield generally similar results, albeit statistically weaker, noting the lower values for true-positive ChEMBLSOM nodes and true-positive ChEMBL compounds withing these nodes. Within the 984 ChEMBL compounds having enriched 6-mers, 434 (44%) share enhanced chemosensitivity (17% with perfect data). This result represents a strong hit rate for associating structure (e.g., 2D fragment composition) with chemosensitivity (i.e., IC50). Retrieving the ChEMBL IC50 values for these hits followed by identifying the median rank of IC50 values within chEMBLSOM projections finds an average median rank of 29%, with the lowest average median rank of 22% for TKL. Collectively, these results indicate that rankings based only on branch-selective chemosensitivity of IC50 appear in the upper 3rd of all values (Table 2).
| Fragments | #ChEMBL with 6-mers | #tp SOM nodes | #tp ChEMBL cmpds (perfect) | p-value | #tp SOM nodes | #tp ChEMBL cmpds(noisy) | p-value |
|---|---|---|---|---|---|---|---|
| AGC | 45 | 5 | 13 5,17,42,992 | 2.01E-03 | 12 | 22 12,79,35,930 | 3.41E-04 |
| CAMK | 86 | 4 | 5 4,32,47,973 | 9.07E-02 | 12 | 54 12,133,39,872 | 3.67E-02 |
| CK1 | 40 | 1 | 1 1,10,9,1036 | 9.98E-02 | 3 | 4 3,47,7,999 |
9.51E-03 |
| CMGC | 263 | 23 | 59 23,73,75,885 | 5.24E-06 | 54 | 97 54,319,44,639 | 2.08E-05 |
| STE | 79 | 3 | 3 3,32,7,1014 | 3.40E-03 | 5 | 5 5,144,5,902 | 7.31E-03 |
| TK | 247 | 28 | 49 28,84,132,812 | 2.71E-03 | 82 | 133 82,304,78,592 | 2.66E-05 |
| TKL | 224 | 17 | 41 17,65,69,905 | 1.40E-04 | 42 | 119 42,281,44,689 | 1.64E-04 |
| Total | 984 | - | 171/984=0.173 | - | - | 434/984=0.441 | - |
| -0.08 |
Note: CAMK: Calmodulin/calcium regulated Kinases; CK: Casein Kinase; TK: Tyrosine Kinase; TKL: Tyrosine Kinase-Like; SOM: Self Organizing Maps
Table 2: Fisher’s exact results for co-occurrence of SOMChEMBL nodes with ligands having branch-chemoselective fragments and SOMChEMBL nodes with branch-chemoselectivity. Colum 1: branch, Column 2: #Chembl compounds with branch-specific 6-mers, Column 3: # true positive SOM nodes (perfect data), Column 4: # true positive ChEMB compounds (perfect data) in true positive SOM nodes, Column 5: p-value for Fisher’s exact testing (perfect data). Columns 6-8 list the values corresponding to Columns 3-5 when using noise-contaminated data. Columns 4 and 7 also list the contingency matrix values (tp,fp,fn,tn) for Fisher’s exact testing. Yellow highlighting indicates TP counts. Green highlighting indicates the count of ChEMBL compounds in true positive chemblSOM nodes.
Thirty-two (32) of the true positive ChEMBL compounds (7% 32/434) share exact 2D matches with KLIFS ligands. The intersection of these ChEMBL compounds with KLIFS data finds 93 PKs (71 for perfect data). The KLIFS records for the 32 matching ChEMBL ligands are lsited. Evident from this table is the appearance of KLIFS ligands bound to numerous PKs; with 13 having two or more binding partners (STI:13, 1N1:13, DB8:11, VGH:9, TAK:5, P06:4, FB8:4, GUI:3, 6QB:3, 6GY:3, T3C:2, 3FE:2 and QUN:2). These 32 matching ligands are associated with 42 PDB PKs, with 18 of them appearing more than once (AKL:11, ABL1:8, CKD2:6, SRC:5, LCK:4, Erk2:4, BRAF:4, MST2:3, EGFR:3, DDR1:3, CHK2:3, TAK1:2, p38a:2, LOK:2, Erbb3:2, EphA2:2, CDC2:2, BTK:2 and ALK2:2). Instances of multiple targets paired with multiple ligands find nearly all to involve PKs in the same kinome branch. For example, 12 of the 13 PKs for STI, 10 of the 13 PKs for 1N1 and 9 of the 11 PKs for DB8 are in the TK branch. In general, ligands targeting multiple PKs in the non-TK branches all involve within-branch partners.
Recall that KLIFS data provides PKs and ligand pairs, while the ChEMBL IC50 data measures IC50 for 461 PKs for >22k ChEMBL compounds. Merging independently constructed branch-specific 6-mers and independently constructed ChEMBL compounds with branch-specific chemosensitivity finds 32 2D-matching ligands that are known to co-crystallize with 42 important oncologic PKs. Co-occurrence of exact 2D KLIFS matches, that also pass the Fisher’s exact enrichment test, appears to be rare, statistically significant and yield exact matches to known ligands. It is difficult to assess the strength of these results for applications to other public databases. However, their existence offers support for this design to yield true hits. Of equal importance are the cases without exact matching PKs. Here there are 402 (434-32) non-structurally matching PKs ligands as candidates for further studies of kinome branch-targeting compounds.
ChEMBL compounds with enriched 6-mers and branch-chemosensitity can also be evaluated for IC50 potency within the original dataset of >22k IC50 values. Figure 13 displays a histogram of average IC50 values for the true positive ChEMBL compounds for each kinome branch. Seven histogram bars are displayed for each kinome branch, where bar height in each group represents the kinome group average IC50 for the true positive ChEMBL compounds, with each bar color-coded according to kinome branch. The asterisks identify the IC50 values within each kinome branch. Inspection finds that AGC, CMGC, STE, TK and TKL have their highest average IC50 values within their respective kinome branch. The exception is for CAMK which is ranked as the 3rd highest average IC50. These results complete the loop, starting from data, to analysis and back to starting data, yielding results that support the overall design (Figure 13).

Figure 13: Histograms of the average IC50 responses for the true positive ChEMBL compounds. The IC50 values for true positive ChEMBL hits are
grouped by kinome branch and used to generate an average IC50 response. The seven sets of histogram bars represent the kinome branch averaged
IC50 for each set of true positive ChEMBL hits. As an example, there are 22 true positive ChEMBL hits for AGC. The average IC50 responses of
these 22 ChEMBL compounds for each kinome branch are, ordered from high to low, AGC (red), CAMK (yellow), TK (purple), CK1 (green), STE
(blue) and CMGC (teal). Absence of a histogram bar indicates no IC50 values. 
FBDD has already achieved noteworthy success. Obstacles and challenges remain when researching goals aimed at enhancing the potential of FBDD. Auxiliary tools, such as proposed here, aimed at discovering ligands that selectively target PKs, offer additional ways to explore applications of FDBB. The primary goal of this work has been to dissect PKs ligands (KLIFS) into fragments (RCDK) and, where possible, assign sets of fragments (-mers) to branch selective PKs (Fisher’s exact testing). This step is followed by screening and statistical testing of bioactivity databases (ChEMBL) for ligands with branch-selective fragments that also exhibit branch-selective chemoselectivity (IC50). The details of this analysis include fragment generation, fragment surveys, fragment enumeration, and fragment enrichment testing for kinome branch selectivity. SOMs were used, separately, to cluster enriched fragment and branch-selective bioactivity data and provide meta-clades for assisting in cataloging the results. Each step in this process was aided by statistical measures for observed results. In summary, the auxiliary tools provided here extend confidence in applications of FDBB for discovering ligands that selectively target PKs in kinome branches.
An application of these results can be used to screen additional databases. For example, enriched 6-mers can be used to mine the Abbott legacy screening dataset published in 2011 [49]. Abbott screened 1497 compounds against 172 protein targets. Filtering (average (IC50)>5.5) and selecting protein targets with a coefficient of IC50 variation above 0.1, reduced this data to 1056 Abbott compounds tested against 156 protein targets. Thirty-three of these filtered Abbott compounds have enriched KLIFS 6-mers, yielding an apparent hit rate of only 3%. However, applying more stringent filtering (average (IC50)>6.6) with a target IC50 standard deviation above 0.7, finds only 37 compounds. Thirty-three compounds are shared in the KLIFS and Abbott structures. Intersection of these sets finds 10 compounds in common; to yield a hit rate of nearly 30%. Parsing the activity values within this intersection finds 28 protein targets comprised of twenty-three proteins in the TK branch (ABL1, ACK1, ALK, AXL, BLK, CSF1R, EGFR, ERBB2, ERBB4, FGFR1, FLT1, FLT3, FRK, IGF1R, JAK2, KDR, LCK, LRRK2, LTK, LYN, PDGFRB, RET and SRC), two in the CAMK branch (PRKCN and PRKAA1), one in the STE branch (MAP4K5), one in the CMGC branch (CLK4) and one in the TKL branch (ACVR1).
An apparent anomaly, notably for cases where the same ligand co-crystallizes with PKs across different kinome branches is displayed. Examples include; ligand 1N1 which co-crystallizes with nine PKs in the TK branch, two in the STE branch and one in the CMGC branch, ligand 3FE (one each in the CAMK and STE branches), ligand DB8 (seven in the TK branch, two in the STE branch and one in the CAMK branch), ligand GUI (two in the TK branch and one in the CAMK branch), ligand STI (twelve in the TK branch and one in the CMGC branch) and ligand TAK (three in the TKL branch and one in the CAMK branch). While these results make assignments of branch selectivity difficult, based only on KLIFS structural data, the enriched 6-mers offer a resolution. Recall that the enriched 6-mers are specific for each kinome branch and a requirement for screening of ligands associated with a specific kinome branch is the presence of at least one enriched 6-mer. For all the ligands cited above, the anomalous entries do not possess branch-specific enriched 6-mers. In other words, co-crystallizations outside the majority kinome branch for each case can be excluded based on their lack of statistical support for enriched branch-selective 6-mers [50].
Cases exist where enriched branch-selective 6-mers are shared across kinome branches. The 6-mer containing fragment N-(4-piperazin-1-ylphenyl)pyrimidin-2-amine (fragment 1513) is enriched in both the TKL and CMGC branches. There are six TKL and six CMGC KLIFS complexes with ligands having fragment 1513. Figure 14 displays the 2D image for these ligands, where fragment 1513 is highlighted in green. Examination of these ligands finds that diverse structures have been appended to the base fragment 1513. Ligand binding site to target Amino Acid (AA) projections of fragment 1513 for these 12 structures are displayed [51-55]. Fragment 1513 is composed of fragment 13 (piperazine) and fragment 257 (N-phenylpyrimidin-2-amine). PKs AAs nearest (5 Å) to fragment 257 consist mainly of hydrophobic residues, interspersed with non-hydrophobic residues, while fragment 13 is mainly solvent exposed. Taking TKL:ALK2:5JUX as an example; KLIFS analysis finds hydrophobic interactions for VAL214, VAL222, ALA233, LEU263, THR283, HID284, TYR285, HID286, GLU287, MET288, GLY289, LEU343 and ALA353, with key interactions in the hinge region provided by TYR285(aromatic) and HID286(H-bond donor). Variations of this general theme are repeated in the other eleven structures in this set, all exhibiting key hydrophobic interactions with fragment 1513, inclusive of aromatic and H-bond donor and acceptor interactions in the hinge region. Evident from this example is the limitation that enriched 6-mers. While capable of excluding most of the kinome branches, in this example, it does not distinguish between ligands targeting either the TKL or CMGC branch (Figure 14).

Figure 14: 2D renderings of 6 ligands that target PKs in the TKL branch and 6 ligands that target PKs in the CMGC branch. Each ligand contains fragment 1513 (shown in green).
An alternative analysis, focused on the base fragment 1513, indicates that this fragment, alone, has the capacity to position itself adjacent to key AAs in the binding site. In support of this claim, Figure 15 displays the superposition of fragment 1513 for all PKs in this set. The average Root Mean Square Deviation (RMSD) for this superposition is 0.523 ± 0.06 Å. The average RMSD for superposition for the TKL and CMGC ligands is 1.273 ± 0.250 Å. The lower panels display the superpositions for the each set of ligands (left: TKL, right: CMGC). Superpositions RMSD for the TKL and CMGC ligands are 1.18 ± 0.49 and 1.42 ± 0.29, respectively. These results find the RMSD for overlapping atoms associated with fragment 1513 to be 2-fold better when compared the complete ligand (Figure 15).

Figure 15: Top panel displays the ball and stick superposition of fragment 1513 for the twelve structures displayed. Fragments targeting the TK and CMGC kinomes have their carbons colored green and black, respectively. Bottom two panels depict the respective superpositions for the complete ligands targeting the TKL kinome (left) and CMGC kinome (right).
Eight FDA approved ligands within the KLIFS database are listed. These are Brigatinib:6GY (FDA approved for ALK-positive metastatic NSCLC), Bosutinib:DB8 (potent TK inhibitor used to treat chronic myeloid leukemia), Dabrafenib:P06 (FDA approved for targeting V600E BRAF), NVP-tae-684:GUI (FDA approved ALK inhibitor), Imatinib:STI (FDA approved to treat Chronic Myelogenous Leukemia (CML), Gastrointestinal Stromal Tumors (GISTs), Dermatofibrosarcoma Protuberans (DFSP), Myelodysplastic/Myeloproliferative Diseases (MDS/MPD), and aggressive systemic mastocytosis, Dorsomorphin:TAK (inhibitor of Bone Morphogenetic Protein (BMP)) signalling, causing cancer initiating cells to lose some stem-cell-like features), Crizotinib:VGH (FDA approved for the treatment of patients with metastatic Non-Small Cell Lung Cancer (NSCLC) whose tumors are anaplastic lymphoma. FDA approved for treating relapsed or refractory, systemic Anaplastic Large Cell Lymphoma (ALCL) that is ALK-positive), AZD5438:FB8 (Inhibitor of CDK1, 2, and 9, enhances the radiosensitivity of Non-Small Cell Lung Carcinoma (NSCLC) cells (Raghavan P, PMC3623267) and Dabrafenib:P06, a selective inhibitor of mutated forms of BRAF kinase.
Table 3 lists the ChEMBL ID, ligand name, 2D structure (highlighted according to its enriched fragments) and their IUPAC names for these eight approved compounds. These FDA hits provide an opportunity to apply 2D fragments for substructure searching. For example, Chembl601719 (VGH:crizotinib) has 9 fragments within its set of enriched 6-mers. The parent VGH structure represents the linkage of 6-mer fragment 216 (4-pyrazol-1-ylpiperidine) and fragment 2520 (3-(phenylmethoxy)pyridine). These linked fragments appear as fragment 3962 within VGH’s set of 9 fragments. The residues within 5 Å of both fragments 216 and 2520 include hydrophobic interactions with LEU1122, VAL1130, ALA1148, LEU1198, MET1199, ALA1200, GLY1202, and ASP1203. Hydrophobic interactions exclusive to fragment 2520 include LYS1150, LEU1196, ARG1253, ASN1254, GLY1269 and ASP1270. Key hinge region H-bond interactions with MET1199 are provided by both fragments, whereas fragment 2520 alone has an H-bond interaction with GLU1197. Fragments 216 and 2520 are positioned in these images as exact atom matches to VGH. A fragment-selective search of the KLIFS ligands with enriched 6-mers using fragments 216 and 2520 finds only the set of PKs for VGH are listed. This result is an indication that fragment-selective searches of other databases, using only fragments 216 and 2520, may yield candidate ligands structurally like VGH (Table 3).
| ChEMBL | PDB | Ligand | Green | Orange | Cyan |
|---|---|---|---|---|---|
| 601719 | VGH:Crizotinib | 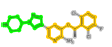 |
216 (4-pyrazol-1-ylpiperidine) | 2520 (3-(phenylmethoxy)pyridine) | - |
| 941 | STI:Imatinib |  |
13 (Piperazine) | 459 (N-(phenylmethyl)aniline) | 2387 (4-pyridin-3-ylpyrimidine) |
| 488436 | FB8:AZD5438 | 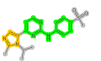 |
257 (N-phenylpyrimidin-2-amine) | 57 (1H-imidazole) | - |
| 2028663 | P06:Dabrafenib | 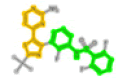 |
3403 (N-phenylsulfanylaniline) | 1267 (5-pyrimidin-4-yl-1,3-thiazole) | - |
| 288441 | DB8:Bosutinib | 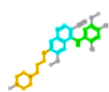 |
520 (N-phenylquinolin-4-amine) | 518(7-(3-piperazin-1-ylpropoxy)quinoline) | 517(Quinoline) |
| 3545311 | 6GY:Brigatinib |  |
374 (N,N’-di(phenyl)pyrimidine-2,4-diamine) | 759 (1-piperidin-4-ylpiperazine) | - |
| 478629 | TAK:Dorsomorphin | 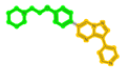 |
500 (1-(2-phenoxyethyl)piperidine) | 502 (3-pyridin-4-ylpyrazolo[1,5-a]pyrimidine) | - |
| 509032 | GUI:TAE684 | 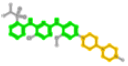 |
374 (2-N,4-N-diphenylpyrimidine-2,4-diamine) | 759 (1-piperidin-4-ylpiperazine) | 257 (N-phenylpyrimidin-2-amine) Hidden by 374 |
| 1421 | 1N1 | 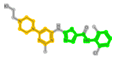 |
1573 (N-(1,3-thiazol-5-ylmethyl)aniline) | 232 (4-piperazin-1-ylpyrimidine) | - |
| 1229592 | 0UN | 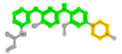 |
506 (4-phenoxy-N-phenylpyrimidin-2-amine) | 13 (piperazine) | - |
Note: PDB: Protein Data Bank
Table 3: Listing of the FDA approved ligands that pass the Fisher’s exact testing for co-occurrence of branch-selective 6-mers and branch-selective chemosensitivity. ChEMBL ID (column 1), PDB ligand name (column 2), 2D rendering of ligand colored according to its component fragments (column 3), fragment index and IUPAC name (columns 4 and higher). Note that in GUI: TAE684 fragment 257 (N-phenylpyrimidin-2-amine) is hidden by 374 (2-N,4-N-diphenylpyrimidine-2,4-diamine).
Visual comparisons of complete ligands and component fragments are displayed below. For example, Figure 16 displays in the left panel the nearest AAs from the PDB target 2xp2, based in the complete ligand VGH (Crizotinib). The right panel displays the nearest AAs to the component fragments (26 and 2520). Here, except for the H-bonding interaction with GLU1197, the AAs surrounding the ligand and fragments are essentially the same (Figure 16).

Figure 16: 2D projections of the binding site pocket for PKs 2xp2. AAs within 5 Å of fragments 216 and 2520 are displayed as residue name and position in the target’s sequence. Colors specify the type of AA (green:hydrophobic, blue:basic, red:acidic). The ribbon around the ligand and fragments represents the 2xp2 target surface, colored by nearest AA. Fragments 216 and 2520 are positioned in the binding site according to exact atomic matches with the complete ligand. Images are rendered using Maestro (Schrodinger Release 2022-3: Schrodinger, LLC, New York, NY, 2021). Views represent 2D projections of 3D images, where each perspective is internally adjusted to uniformly display AAs. As a result, the location of binding site AAs for each ligand will depend on the perspective.
A similar analysis of Chembl941(STI: Imatinib) finds 13 KLIFS structures, 5 for ALB1, 1 for ABL2 , DDR1, FMS, KIT, LCK, PDGFRa and SYK in the TK branch and one case for p38a in the CMGC branch. The latter case can be excluded based on lacking enriched 6-mers in the CMGC branch. STI has 14 fragments within its list of enriched 6-mers. There are various combinations of fragments that can be selected as representatives of STI. The following example links fragment 13 (piperazine), fragment 459 (N-(phenylmethyl) aniline) and fragment 2387 (4-pyridin-3-ylpyrimidine). These linked fragments appear as 2699 within STI’s set of 14 fragments [56].
The residues within 5 Å of STI in PDB:2hyy:A:STI include hydrophobic interactions with LEU248, TYR253, VAL256, ALA269, VAL270, LYS271, GLU286, VAL289, MET290, VAL299, ILE313, THR315, PHE317, MET318, GLY321, PHE359, ILE360, HID361, ARG362, LEU370, ALA380, ASP381 and PHE382. Hinge region interactions include MET318 (H-bond) and PHE317 (aromatic). Interactions shared between fragments 459 and 2387 include VAL256, ALA269, LYS271, VAL299, THR315, LEU370, ALA380 and PHE382. Interactions shared between fragments 2387 and 13 include GLU286, VAL289, ILE293, LEU354 and HID361. Hinge region interactions are provided by fragment 2387 (PHE317: aromatic and MET318: H-bond). Fragments 13, 459 and 2387 are positioned in these images as exact atom matches to STI. A fragment-selective search for KLIFS ligands possessing frags 13, 459 and 2387 identifies the PKs are listed for STI. In addition this fragment-selective search also yields the ligand MPZ which co-crystallizes with the PKs SRC in the TK branch (TK: SRC: 1y57: A: MPZ). Although the ligand MPZ does not have an exact match in the ChEMBL dataset, fragment-selective mining would indicate a potential role in PKs inhibition comparable to STI.
The ligand DB8 Bosutinib targets 10 members of the TK branch and one member of the STE branch. DB8 consists of three overlapping fragments; 517 (quinoline), 518(7-(3-piperazin-1-ylpropoxy) quinoline) and 520(N-phenylquinolin-4-amine), jointly represented as fragment 519. The residues in PKs 3ue4, within 5 Å of the DB8 fragment, finds hydrophobic interactions with LEU248, VAL256, ALA269, VAL270, LYS271, MET290, VAL299, ILE313, ILE314, THR315, PHE317, MET318, THR319, TYR320, GLY321, LEU370, ALA380 and PHE382. Fragment 519 is positioned in these images as exact atom matches to DB8. A database search of these three substructures identifies the PKs are listed for DB8, with the inclusion of the ligand XZN, which targets LOK in the STE branch STE:LOK:4bc6:A:XZN. These fragments appear as enriched 6-mers in the TK and STE branches. This result is an indication that DB8 and XZN share fragments, with structures sharing these fragments having activity in the TK and STE branches.
The P06 ligand is Dabrafenib (ChEMBL2028663). There are 9 fragments associated with P06, with fragment 3403 (N-phenylsulfanylaniline) and fragment 1267 (5-pyrimidin-4-yl-1,3-thiazole) representing most of the ligand. Using the PKs of 4xv2, there are hydrophobic interactions with ILE463, GLY464, SER465, GLY466, VAL471, ALA481, LYS483, LEU505, LEU514, LEU515, PHE516, ILE527, THR529, GLN530, TRP531, HID532, PHE583, GLY593, ASP594 and PHE595. Here the hinge region interactions are CYS532 (H-bond) and TRP531 (aromatic). Within this set, fragment-selective searching with 3403 and 1267 identifies the four ligands associated with P06, with the inclusion of TKL:RIPK2:5ar8:A:XYW. Ligand XYW does not appear because it does not have an exact ChEMBL match. However, its shared substructure with P06 would also suggest the PKs of RIPK2 in the TKL branch for ligands with these fragments.
The KLIFS ligand FB8 is the FDA approved drug AZD5438 (ChEMBL488436). The structure of FB8 is comprised of four overlapping fragments: 1377 (4-(1H-imidazol-5-yl)-N-phenylpyrimidin-2-amine), 1279 (4-(1H-imidazol-5-yl)pyrimidine), 257 (N-phenylpyrimidin-2-amine) and 57 ((1H-imidazole). Fragment-selective searching yields the ligands reported and the additional ligands IM9, FRT, I19 and 4WE for targeting CDK2 in the CMGC branch. None of these ligands have exact ChEMBL matches.
The KLIFS ligand 1N1 (ChEMBL1421) is not an FDA approved drug, however there are ten KLIFS PKs for 1N1 are listed. The key fragments in the 1N1 structure are 1573 (N-(1,3-thiazol-5-ylmethyl)aniline) and 232 (4-piperazin-1-ylpyrimidine). Fragment-selective searching yields the ligands reported with the addition of the ligand 4B7, which also is co-crystallized with ABL1 in PKs 4yc8. This result suggests a novel candidate ligand for targeting ABL1.
Ligand F3Z (ChEMBL 3590107) co-crystallizes with CMGC:Erk2:6gdm:A:F3Z. F3Z is composed of 3 IUPAC fragments 2139 (3-pyridin-4-yl-1H-indazole), 1662 (2-phenylpyrimidine) and 1998 (1-(2-pyrrolidin-1-ylethyl)piperazine). These 3 fragments, in combination with 12 other fragments (1,9,13,44,93,121,226,1662,1988,1993,1995,1998,2136,2139,2141), each comprised of different portions of these three IUPAC named fragments, appear in the enriched 6-mers only for the CMGC kinome branch. Fragment-selective searching, using 1662, 1998 and 2139, finds that CMGC selectivity also exists for KLIFS ligands FOE (CMGC:Erk2:6gjd:A:F0E ) and NOV (CMGC:Erk2:6opi:A:N0V), neither of which have exact structural matches to the >22k ChEMBL ligands. F3Z and FOE are the only KLIFS ligands that share fragments 2139, 1662 and 1998, whereas NOV shares fragments 2139 and 1662. These three ligands selectively target Erk2 in the CMGC kinome branch [57].
Table 4 summarizes the surface area results for the complete ligand and the component fragments of the examples reported above. This table lists the target PKs (column 1), the designation of ligand or fragment (column 2), unbound surface area (column 3) and bound surface area (column 4), the difference of bound and unbound surface area (column 5) and the fraction of surface lost in the bound state (column 6). These results indicate that greater than 80% of the complete ligand’s bound surface area is due to its component fragments (Table 4).
| PDB | Target/ligand | Unbound SA | Bound SA | Delta | Delta/unbound SA |
|---|---|---|---|---|---|
| 3ue4 | ABL1 | 691.79 | 216.44 | 475.36 | 0.69 |
| 3ue4 | DB8 | 826.25 | 240.44 | 585.81 | 0.71 |
| 2xp2 | ALK | 619.97 | 135.86 | 484.11 | 0.78 |
| 2xp2 | VGH | 683.97 | 134.59 | 549.38 | 0.8 |
| 2hyy | ABL1 | 782.04 | 63.08 | 718.97 | 0.92 |
| 2hyy | STI | 828.17 | 73.92 | 754.25 | 0.91 |
| 4xv2 | BRAF | 599.29 | 39.44 | 559.85 | 0.93 |
| 4xv2 | P06 | 737.19 | 62.25 | 674.94 | 0.91 |
Note: PDB: Protein Data Bank
Table 4: Surface area summary for the PKs discussed above. Target PKs (column 1), designation of target PK or its ligand (column 2), unbound surface area (column 3) and bound surface area (column 4), the difference of bound and unbound surface area (column 5) and the fraction of surface lost in the bound state (column 6). Results for each PDB are listed as consecutive rows with the same highlighting.
The preceding examples illustrate a means to integrate a coarse-grained 2D fragment approach, combined with chemosensitivity data and applications of Fisher’s exact testing to yield results that mesh with crystallographic structural details. A widely used component of FBDD involves in silico ligand docking to screen for candidate PKs. As with FBDD, in silico docking is a vital part of the overall task of drug discovery. Citing the above example for the VGH ligand with nine KLIFS complexes, seven for ALK, one for MET and one for ROS, all specifically target the TK branch. Superposition of these ligand x-ray structures finds that the VGH:ALK structures have Root Mean Square Deviations (RMSDs) of less than 0.1 Å, while VGH:MET and VGH:ROS deviate from the VGH:ALK set by greater than 1.6 Å. The rmsd of VGH:MET and VGH:ROS is 1.32 Å. These results are consistent with the KLIFS Internal Profile Fingerprint similarity (IPF) within the VGH:ALK set of greater than 0.9, while the IPF similarity score drops to 0.64 and 0.62 for VGH:MET and VGH:ROS, respectively. Although not done here, a docking search for PK targets using a VGH:ALK conformer may not have identified the MET and ROS targets. Whereas the coarse-grained approach using 2D fragments, combined with chemosensitivity data and applications of Fisher’s exact testing, includes VGH within the list of candidate ligands [58].
Although these results represent preliminary extensions of this analysis, two points for future use can be proposed. First, crystallographic ligands that target PKs across different kinome branches appear, in the above cases, to be rejected based on their lack of branch-specific enriched 6-mers. Contributing to this result is, in part, due to the failure of the Fisher’s exact test to yield a significant result when fragments are shared across kinome branches. As a cautionary note, recall that the best case results for using -mers to retrospectively identify correct cases found 6-mers, with 84% success and 16% failure rates. Clearly, utilization of 6-mers for compound screening, while having a relatively high success rate, has a non-trivial likelihood for failure. Second, utilization of kinome branch-selective 6-mers appears capable of providing pharmacophore-based screening strategies. Third, necessary components of this analysis include, at a minimum; the selection of input databases, filtering thresholds for input data, clustering methodologies and variations in statistical methods. The balance of these components, as reported here, represents one choice. Validation of this and other choices require dealing with many options. However, validative assessments will benefit most from applying these choices, proposed in the design reported here, to the continually increasing quantity of input data.
Decomposition of PKs ligands into fragments, followed by statistical testing for fragment enrichment within separate kinome branches, provides an effective means for identifying kinome-selective fragment subsets. Associating sets of enriched branch-selective fragments with ligands also possessing branch-selective chemosensitivity is rare however the co-occurrence of exact 2D KLIFS matches, that also pass the Fisher’s exact enrichment test, yields exact matches to FDA approved ligands. In general, fragment composition is an effective means for probing chemical databases for candidate compounds that selectively target PKs within kinome branches.
This project was funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contracts No. HHSN261200800001E and HHSN261201700007I. There was no additional external funding received for this study.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Covell DG (2023) Bioinformatic Analysis: Linking Chemosensitivity and 2D Structural Features of Ligands Targeting the Protein Kinases in Branches of the Kinome Tree. Drug Des. 12:244.
Received: 01-May-2023, Manuscript No. DDO-23-24013; Editor assigned: 03-May-2023, Pre QC No. DDO-23-24013 (PQ); Reviewed: 17-May-2023, QC No. DDO-23-24013; Revised: 24-May-2023, Manuscript No. DDO-23-24013 (R); Published: 02-Jun-2023 , DOI: 10.35248/2169-0138.23.12.244
Copyright: © 2023 Covell DG. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.