Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2019)Volume 8, Issue 2
The hepatic microsomal ethanol-oxidizing system (MEOS) was initially confronted with much uncertainty, skepticism, scientific antagonism, and heavy discussions. Viewed as scientific challenges, this stimulated further research, and led to its successful separation from both, alcohol dehydrogenase and catalase, and its reconstitution that allowed defining the individual components of MEOS: cytochrome P450 (CYP), reductase, and phospholipids. Subsequently, it was challenging to elucidate the molecular basis of the microsomal ethanol oxidation. Unlike a usual dehydrogenation or simple oxidation process, ethanol oxidation via MEOS proceeds via reactive intermediates, commonly known as reactive oxygen species (ROS) and generated by various microsomal CYP isoenzymes including CYP 2E1, all of which are established components of MEOS. Due to its radical scavenging properties, ethanol combines with a small fraction of hydroxyl radicals and undergoes oxidation while the remaining radicals attack phospholipids of liver cell membranes. Chronic alcohol use enhances MEOS activity by upregulating CYP 2E1 combined with ROS generation, and thereby increases the metabolism of ethanol to acetaldehyde, its first metabolite with a high hepatotoxic potential. Considering the involvement of various CYP isoenzymes as constituents, MEOS is now best defined as a multi-CYP isoenzyme system, participating in ethanol metabolism and responsible for the molecular-based alcoholic liver disease.
Microsomal ethanol-oxidizing system; MEOS; Cytochrome P450 2E1; CYP2E1; Reactive oxygen species; ROS
With its short chemical chain, alcohol (syn. ethyl alcohol, ethanol, or in short EtOH) attracts continuous interest in molecular sciences as illustrated by two processes, one of these molecular considerations relates to the production of alcohol and the other one focuses on its degradation [1-6]. The first molecular aspects are to be viewed regarding alcohol as a nature-based product derived from plants that provide glucose (C6H12O6) as end product via photosynthesis in their chloroplast, whereby glucose in turn becomes the starting product of EtOH (C2H5OH) [1,2]. For instance, during wine production, glucose of grapes is split by fermentation into 2 pyruvate (C3H5O3), and the 2 pyruvate are converted to 2 acetaldehyde (C2H4O). In a final step, alcohol dehydrogenase (ADH), contained in baker’s yeast and added by the wine grower to the grape juice, helps produce 2 EtOH from 2 acetaldehyde, using 2 NADH + H+ as coenzyme, whereby NADH is the short form of reduced nicotinamide adenosine dinucleotide [3,4]. Interestingly, these enzyme–based biological steps of alcohol production as facilitated by the wine grower may partially be reverted in humans, who consume the EtOH contained in the wine. Such events in humans in turn will lead to the second aspect of molecular interests.
These other processes in humans occur in their livers, where the alcohol molecules meet other molecules and enzymes known for their capacity of degrading EtOH to acetaldehyde:
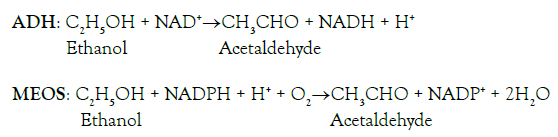
For the metabolism of EtOH, preference is given to two enzymes, namely to ADH, which requires NAD + H+ as cofactor and reverts the enzymatic reaction of ADH during the wine production, and also to the microsomal ethanol-oxidizing system (MEOS), which is dependent on reduced NADPH + H+ (nicotinamide adenosine dinucleotide phosphate), whereas little if any alcohol is degraded by catalase [5]. Many molecular questions relating to MEOS and other issues have been studied in humans as well as in animals, using specific experimental animal models [7-9]. Current alcohol research on MEOS focuses on the highlights of its characteristics including its nature and role promoting the microsomal oxidative stress via cytochrome P450 (CYP) and especially its isoenzyme CYP 2E1, a major constituent of MEOS [5]. Many other new aspects merit further consideration, such as the circadian rhythms of MEOS activity and liver injury, and mechanistic aspects of the intestinal microbiome as basis for the gut-liver axis, triggering the initiation of experimental alcoholic liver injury and human alcoholic liver disease [5].
The present article critically and comprehensively analyzes molecular aspects of MEOS and presents a broad overview of MEOS characteristics as well as of recent developments of MEOS related to toxicity within the last years.
Data search and sources
The PubMed database was used to identify publications for the following terms: microsomal ethanol-oxidizing system, MEOS, cytochrome P450 2E1, and CYP 2E1. Publications from each searched segment were analyzed. The search was completed on 12 July 2019. For the search term microsomal ethanol-oxidizing system overall 135,000 hits were obtained, for MEOS 4,740,000 hits, for cytochrome P450 2E1 324,000 hits, and for CYP 2E1 1,330,000 hits. For the reference list, respective reports had to be selected on basis of relevance for the topic. Articles in English language were preferred.
The data source is also a large and actualized private scientific archive, which contains original full-length publications of MEOS and CYP 2E1. Prior to the final analysis, the publications were assessed regarding quality and data completeness. The final compilation consisted of original publications, consensus reports, and review articles, with inclusion of the most relevant ones in the reference list of this review.
Alcohol, drugs and the hepatic endoplasmic reticulum
Until the middle of the eighties, many drugs and several exogenous chemicals were known as substrates of the hepatic endoplasmic reticulum, which can be obtained from liver homogenates as microsomal fraction upon several centrifugation steps including ultracentrifugation. Most of these substrates can also cause a striking proliferation of the endoplasmic reticulum as indication of some metabolic interactions or even enzymatic degradation. However, the topic of EtOH as potential substrate for the microsomal drug-metabolizing enzymes was neglected, likely due the general assumption that the hepatic ADH metabolizes all of the ingested ethanol, leaving no room for another enzyme. Conditions changed with the observation that prolonged alcohol use causes proliferation of the endoplasmic reticulum in the liver cells [10], leading to the proposal that liver microsomes can indeed metabolize EtOH [11,12], as summarized recently [5].
Discovery of MEOS
Published first in Science in 1968 [11] and subsequently in the Journal of Biological Chemistry in 1970 [12], Lieber and DeCarli described EtOH as a new substrate of hepatic microsomes and named their discovered innovative enzyme reaction the microsomal ethanol-oxidizing system (MEOS) [11,12]. Apart from animals, MEOS activity was found also in the liver of humans, which opened the view of its potential importance in patients with alcoholic liver disease. As expected, their discovery was early recognized as a cornerstone and significant scientific breakthrough in the field of clinical and experimental alcohol research and led to many publications around the world within the last 50 years [5], starting with the original publications [11,12].
Explicitly named as system rather than as microsomal ethanol oxidase, MEOS was assumed as a reaction not based on a single enzyme but resembling more a drug metabolizing system, consisting at least of cytochrome P450 (CYP) and the corresponding reductase [11,12] in a similar way using benzphetamine as another substrate described by Lu and Coon in 1968 [13]. In line with the microsomal drug metabolizing systems also called the mixed function oxidase, MEOS required molecular oxygen and NADPH as cofactor for its activity, was not active under an atmosphere of nitrogen or with boiled microsomes, and exhibited reduced activities under an atmosphere of CO that binds to CYP [11,12]. These additional characteristic features were highly suggestive of the involvement of CYP in MEOS activity, an assumption that proved correct later on [5,14].
At the time of the first publications on the MEOS discovery, Lieber and DeCarli already communicated that in the microsomal factions they used for the MEOS assay, catalase in small amounts was found as a common contaminant of microsomes, because for their preparation liver homogenates had to be used that contain all cellular components including enzymes [11,12]. Considering the issue of potentially contaminating enzymes that could interfere with the MEOS assay leading to disputable results, studies with inhibitors had been initiated and their results were part of the initial reports in order to clarify the situation [11,12]. These early inhibitor studies showed some inhibition of the microsomal EtOH oxidation, if azide as a known potent catalase inhibitor was added in vitro or if 1,2,4-aminotriazole as a strong catalase inhibitor was applied in vivo prior to the MEOS assay. From the results of these specific inhibitor studies the conclusion was reached and published that liver microsomes contain an enzyme system that works independently from catalase. To be on the safe side and to rule out that contaminating ADH could be a constituent of MEOS, additional inhibitor studies were carried with pyrazole as a potent ADH inhibitor, showing insensitivity of MEOS activity in the presence of in vitro added pyrazole. Combining the catalase and ADH inhibitor studies, sufficient evidence was therefore presented that liver microsomes contain a system capable of oxidizing EtOH not requiring catalase or ADH [11,12]. These early inhibitor studies also suggested that catalase contaminating the microsomal fraction is enzymatically active through both, its catalatic reaction, which decomposes H2O2 to water and O2, and its peroxidatic reaction, which peroxidizes EtOH to acetaldehyde and water.
Consensus ambiguity
Soon after the early publications on MEOS discovery [11,12], several groups found this topic of interest and started with own studies [15-23], in search to reproduce or disregard the results as initially reported [11,12]. First of all and in line with the initial description of MEOS [11,12], the engaged groups confirmed that rat liver microsomes are capable of oxidizing EtOH to acetaldehyde in a reaction requiring NADPH as cofactor [15-23]. They also confirmed that rat liver microsomes contained catalase as contaminant. Therefore and in line with the previous data [11,12], consensus existed on all results of the basic experimental procedures [15-20]. However, the subsequent interpretation of the results became a matter of debate with contrarian consensus among the various opposing groups [15-23]. This outside consensus was summarized in the title of one of their reports: ethanol oxidation by liver microsomes, evidence against a separate and distinct enzyme system [15]. Strong words, indeed, with little change to survive expert clarification by many groups as referenced [5,14].
Initial controversies around the nature of MEOS
Clearly, any discovery in medicine or natural sciences is challenging when published for the first time, conditions that also applied to MEOS. It is often a matter of reputation, funding of research projects, resentment, and annoyance. The easiest way to handle this scientific issue is by declaring the discovery as not real or as the result of an artifact due to confounding variables. In fact, this occurred with MEOS preferentially in the early seventies as expressed in opinions questioning the nature of MEOS and its independence from ADH and catalase [15-23].
Most of the discussions regarding MEOS activity focused more on the claimed role of catalase, which is not recognized as a constituent of microsomal membranes but known merely as a contaminant [15-23]. The opinion of these opposing groups was in sharp contrast to the initial concept presented at the time of MEOS discovery, which suggested that a large fraction of MEOS is due to a distinct enzyme reaction functioning independently from and not requiring catalase [11,12]. The opponents also claimed that the assumed reaction required not only catalase but also H2O2 possibly generated by hepatic microsomes. Under normal conditions and in the intact liver cell, catalase resides in the peroxisomes and is commonly far away from the endoplasmic reticulum where MEOS resides. In the absence of ethanol and likely also in its presence, H2O2 generated within the liver cell is rapidly neutralized by antioxidants and glutathione aiming to remove this potentially cytotoxic reactive oxygen species (ROS).
Solubilization of MEOS
In order to further characterize MEOS, additional studies were required [24-29], which included the topic of substrate specificity [26-28] and focused on column chromatography [24,25,27-29]. In search for its constituents, approaches were initially undertaken to solubilize microsomal membranes that contained MEOS. Previous publications had identified the microsomal constituents of a drugmetabolizing enzyme following effective membrane solubilization and column chromatography, approaches that allowed for retained enzyme activity with the substrate benzphetamine as reported for the microsomal benzphetamine demethylase by Lu and Coon [13]. Retaining enzyme activity following solubilization of membrane structures is often tricky due to the risk of partial or complete inactivation of the enzymes of interest, making enzyme characterization then obsolete. Therefore, the primary aim was to attain gentle solubilization of MEOS with retained enzyme activity, which was achieved with a procedure as described in detail [24]. As a result from the solubilization procedure, MEOS activity was obtained from the solubilized microsomal mixtures at recovery rates of up to 85% of the original activity of intact microsomes, fairly good conditions to be used as starting biological material for subsequent approaches of its isolation or perhaps reconstitution. However, preliminary own approaches using a variety of preparative methods to isolate microsomal constituents such as CYP and the reductase from the mixture of solubilized microsomal membranes remained unsuccessful regarding reconstitution of MEOS activity. The experimental protocol was then shifted to column chromatography as reported by Lu and Coon [13]; at the end and due to the generous advice of Anthony Y. H. Lu, publishable results were obtained [24].
Ion exchange chromatography
Solubilized rat liver microsomes retaining strong MEOS activities were submitted as mixture to ion exchange chromatography using activated DEAE (Diethyl-Amino-Ethyl) cellulose as resin, and elution was achieved using a linear salt gradient of KCl starting with 0 M KCl and ending with 0.5 M KCl [24]. Under the assumption that MEOS could have lost part of its activity during the chromatographic procedure, initial studies focused on the determination of the recovery rate. Compared to the overall MEOS activity applied on top of the column, up to 80% was finally recovered in the eluates depending on the preparative conditions. The partial loss of MEOS activity was due to conversion of the active cytochrome P450 to P420 as its inactive form. Additionally, elution of MEOS may have been incomplete due to strong binding of microsomal constituents to DEAE cellulose impairing their release by KCl. As compared to the linear KCl gradient with up to 0.5 M KCl [24], the use of a stepwise KCl gradient with maximum 0.4 M KCl at the final step provided even better recovery results of MEOS, likely because P420 was less produced from cytochrome P450 due to a speedier elution procedure [25]. This quicker method leading to retained MEOS activity was also reported as reproducible in an affirmative study by Damgaard [29].
Separation from ADH and catalase
Following solubilization of microsomes and during the subsequent ion exchange column chromatography, ADH and catalase activities can effectively be removed from microsomal enzymes that oxidize ethanol, findings supporting the assumption that ADH and catalase are neither closely associated to microsomal enzymes nor constituents of microsomal membranes embedded in the membranes of the endoplasmic reticulum [24,25,27-29]. This ended the emotional discussions with clarifying positions that hepatic microsomes contain a distinct enzyme system, which metabolizes EtOH independently from ADH and catalase. Consequently, these chromatographic but also other supporting studies then presented a few winners [11,12], contrary to others who had heavily opposed for many years and had been proven wrong [15-23].
Isolation of MEOS and its tentative microsomal constituents
The elution pattern of solubilized liver microsomes duly fulfilled clarification regarding the nature of MEOS as a unique enzyme system [24]. It clarified not only the need of a successful physical separation of ADH and catalase from MEOS, but at the same time, it also suggests microsomal components as potential constituents of MEOS. Therefore, a closer look on the elution pattern is warranted (Figure 1).

Figure 1: Purification of MEOS and its isolation from catalase and ADH activities. Separation was achieved by DEAE-cellulose column chromatography after solubilization of liver microsomes obtained from rats fed an ethanol containing liquid diet for 3 weeks. Abbreviations: ADH, Alcohol dehydrogenase; E280 nm represents the protein content of the eluates; MEOS, Microsomal ethanol-oxidizing system. The figure is modified from a figure published in a previous report [24], reproduced with permission of Elsevier.
Initially, the void volume is eluted up to around 220 mL, whereby its elution pattern early shows 3 peaks, the highest one corresponds to the protein curve expressed as E280 nm, below that appears catalase with a left-sided peak, and ADH shows with the lowest, somehow rounded peak (Figure 1). Following the initial elution of ADH and catalase in the void volume and starting with an elution volume at around 330 mL, thereby far away from prior ADH and catalase, all microsomal components begin to appear. Here, the first peak corresponds to cytochrome P450 while the second one represents E280 nm as the protein curve of the eluates, followed by a third small peak with two adjacent small shoulders and by a fourth high peak, these last two peaks representing MEOS activity. At around 770 mL, the reductase peak emerges, which is the NADPH cytochrome P450 reductase measured as NADPH cytochrome c reductase for reasons of convenience, and this is followed by the phospholipid peak at around 790 mL elution volume. Analyzing the elution pattern in detail, the following conclusions may therefore be drawn (Figure 1): (1) MEOS activity was recovered only in fractions containing all three microsomal constituents, namely cytochrome P450, the reductase, and phospholipids, corresponding to eluates between around 330 mL and 830 mL; (2) MEOS activity was not found in fractions containing only the reductase, phospholipids, or both together in the absence of cytochrome P450, corresponding to eluates from around 830 mL to 1080 mL; (3) MEOS activity was not recovered in fractions containing only phospholipids but not cytochrome P450 and not the reductase, corresponding to eluates starting at around 970 mL; and most importantly, (4) MEOS activity was found in eluates between around 330 mL and 830 mL, which were completely free of ADH and catalase activities [24]. The interpretation of these data led to the clearly outlined suggestion that MEOS consists of cytochrome P450, the reductase, and phospholipids [24].
For the chromatographic studies of MEOS isolation, microsomes were used from livers of rats [24,25,28], and mice [27], and the salt gradient for the elution was either a linear one [24] or a stepwise one [25,27,28]. Using our published elution procedure of a stepwise KCl gradient [25], highly appreciated was the subsequent report of Damgaard in 1982 [29], who reaffirmed the validity and reproducibility of our initial results in all details [25]. This reassured our proposed participation of special microsomal enzymes and constituents in MEOS and its independence from ADH and catalase.
Characteristics of the isolated MEOS
Basic features: MEOS as isolated by chromatography is characterized by the following features [24]: (1) the enzymatic reaction of MEOS is dependent on NADPH and molecular oxygen; (2) the pH optimum is in the physiologic range; (3) the reaction is linear with the protein content and abolished by boiling; (4) its activity is insensitive to pyrazole, an ADH inhibitor, and also to azide, a catalase inhibitor, but is reduced by butanol as the first ever described chemical inhibitor of MEOS and is also diminished under an atmosphere of CO. Circumstantial evidence suggests that CYP, the reductase, and phospholipids are constituents of MEOS, but the responsible CYP isoenzyme form(s) could not be determined at the time of isolation, simply because the appropriate techniques were not available. However, the previously published elution pattern reported already that MEOS activity did not parallel the CYP pattern. This finding suggested differences among the CYP activity strength for ethanol with variable MEOS turnover over numbers per CYP molecules, whereby the highest MEOS peak in the later phase of elution was found correlated with low amounts of CYP that was obviously more active metabolizing EtOH compared to CYPs eluted earlier.
Substrate specificity: The isolated MEOS fraction exhibits a broad substrate specificity with respect to short length alcohols, which then led temporarily to an adaptation of terminology because MEOS changed to MAOS (microsomal alcohol-oxidizing system) [24-28]. The substrate specificity can easily be determined by substitution of ethanol in the usual MEOS assay by any other alcohol or any substrate but this may require a specific substrate related analytic approach. Studies on the substrate specificity of the microsomal system are of importance to further differentiate MEOS especially from catalase [24-28], as summarized in a list (Table 1) [5,14,30].
| Characteristics | ADH | MEOS | Catalase |
|---|---|---|---|
| Intracellular localization | Cytosol | Endoplasmic reticulum | Peroxisomes |
| Co-factor | NAD+ | NADPH + H+ | - |
| Co-substrate | - | Molecular oxygen | H2O2 |
| Reaction products | Acetaldehyde NAD + H+ |
Acetaldehyde NADP+, H2O |
Acetaldehyde H2O |
| Kinetics | |||
| Km (ethanol) | 0.5 – 2.0 mM | 7 – 11 mM | 0.6 – 10 mM |
| Km (O2) | - | 8.3 µM | 50 µM |
| pH optimum | 11 | 6.9 – 7.5 | 5.5 |
| Inhibitory effect | |||
| Pyrazole (0.1 mM) | ++++ | 0 | (+) |
| Cyanide (0.1 mM) | N.D. | 0 | ++++ |
| Azide (0.1 mM) | 0 | 0 | ++++ |
| Substrate specificity | |||
| Methanol | ++ | ++ | ++++ |
| Ethanol | +++ | ++++ | ++++ |
| n-Propanol | ++++ | +++ | (+) |
| n-Butanol | ++++ | ++ | 0 |
| n-Pentanol | ++++ | + | 0 |
| i-Propanol | + | + | 0 |
| t-Butanol | 0 | + | 0 |
| Increase in activity following chronic ethanol consumption | 0 | ++++ | 0 |
| Enzyme isolation | + | + | + |
| Isoenzymes | + | + | + |
Table 1: Differentiation between ADH, MEOS and catalase.
Whereas lower alcohols such as methanol and EtOH are substrates for both, the microsomal system and catalase, propanol, butanol, and pentanol are excellent substrates for the microsomal system but not for catalase (Table 1) [24-28]. These additional results clearly provide additional evidence that catalase is not an obligatory or facultative constituent of MEOS and disproved views to the contrary [15-23]. The completion of these early studies on MEOS isolation and substrate specificities [24-28] allowed the focus on many other relevant specificities [24-28] allowed the focus on many other relevant projects aiming to clarify first the microsomal components of MEOS and later the role of CYP 2E1 [5,14].
Drugs as substrates: Eluates containing the isolated MEOS but not catalase also retained drug-metabolizing activities with respect to aminopyrine, benzphetamine, and aniline as substrates, in addition to their oxidation of EtOH [25]. These results affirm that also other chemicals are metabolized by liver microsomal constituents in the absence of catalase and recommend this chromatographic method of enzyme isolation for other drug studies on mechanistic steps, whereby addition of catalase to remove H2O2 could assess, for instance, a potential role of H2O2 in the metabolic drug reactions. Additional studies with the eluates containing the isolated MEOS have earlier shown that the NADPH oxidation rate was substantially higher when assessed in the presence of benzphetamine while virtually unchanged or marginally decreased rates were found with aniline or EtOH added to the incubation medium [25]. Consequently, ethanol itself failed to enhance the NADPH dependent oxidation and may therefore not stimulate its own metabolism via MEOS through generation of reactive intermediates.
Partial purification of microsomal constituents
Notably, if the method of MEOS isolation to separate it from catalase was not used as an initial step [24,25,27,28], problems commonly emerged in other studies reporting partially purified microsomal constituents due to contaminating catalase. Nevertheless and in line with previous suggestions [24], preliminary reconstitution experiments using partially purified microsomal constituents led to the tentative conclusion that MEOS requires CYP and the reductase [31,32], views supported by other reports as comprehensively discussed in several reports [33-35] including in Methods of Enzymology [35].
Successful reconstitution of MEOS with purified microsomal components
Only ten years after the published discovery of MEOS, purified microsomal constituents allowed the complete reconstitution of MEOS, published by Miwa et al. from the group of Anthony Y. Lu as senior author [36]. Based on these results, it was now affirmed that MEOS consists of CYP, the reductase, and phospholipids, findings in line with previous suggestions confirming results of the isolated MEOS published earlier [24] as well as later [25,27-29].
Established microsomal components of MEOS
CYP has early been proposed as constituent of MEOS, but the responsible isoenzyme remained unassessed [24]. It is now clear that its isoenzyme CYP 2E1 is the most active isoenzyme promoting microsomal ethanol oxidation [5,14,37], together with other CYP isoenzymes like CYP 1A2, 2A6, 2B6, 2D6, and A4 (Table 2) [5,37].
| Cytochrome P450 Isoenzyme | MEOS activity/Cytochrome P450 |
|---|---|
| 1A2 | 10.90 |
| 2A6 | 3.75 |
| 2B6 | 2.89 |
| 2D6 | 0.70 |
| 2E1 | 11.51 |
| A4 | 3.38 |
Table 2: MEOS and its cytochrome P450 isoenzymes.
Metabolic variability is a characteristic feature of the various CYP isoenzymes that all together constitute MEOS activity, whereby individual isoenzyme activity is best defined as its turnover number, calculated as MEOS activity in units per CYP isoenzyme unit (Table 2). Therefore, isolated MEOS obtained by chromatography is best defined with the following microsomal constituents: (1) cytochrome P450 with its preferred isoenzyme CYP 2E1 as well as other isoenzymes; reductase in short or expanded as NADPHcytochrome P450 reductase; (3) and microsomal phospholipids (Figure 2) [24].

Figure 2: Constituents of MEOS. Hepatic microsomal cytochrome P450 2E1 and NADPH-cytochrome P450 reductase are obligatory constituents of the microsomal ethanol-oxidizing system (MEOS), the metabolic reaction requires also phospholipids but the site of their reaction is unknown. Reproduced from a previous report [30], with permission of the Publisher Taylor & Francis.
Cytochrome b5, is another hemeprotein and a constituent of the liver microsomal membranes, together with its corresponding cytochrome b5 reductase. During the column chromatographic procedure, these two constituents are eluted in fractions exhibiting MEOS [25,27], confirmed by a subsequent study [29]. However and by definition, MEOS is a system solely of the hemeproteins CYPs [5,14] and not of other hemeproteins such as cytochrome b5, with its corresponding NADH-cytochrome b5 reductase, although interactions between CYP 2E1 and cytochrome b5 with its reductase regarding alcohol metabolism are under discussion and likely occur [14,38].
Tentative molecular events of MEOS activity
In many biological systems with integrated oxidative and reductive molecular processes, metabolic events may be associated with the generation of various forms of reactive oxygen species (ROS), conditions also described for the liver responsible for the microsomal oxidation of EtOH where many toxic radicals with variable chemical structures are produced (Table 3) [5,14,39-60].
| Selected potentially toxic metabolites and reactive O2-species due to hepatic ethanol degradation |
|---|
| Acetaldehyde C2H4O |
| Ethoxy radical CH3CH2O |
| Hydroxyethyl radical CH3C(.)HOH |
| Acetyl radical CH3CHO. |
| Singlet radical 1O2 |
| Superoxide radical HO.2 |
| Hydrogen peroxide H2O2 |
| Hydroxyl radical HO• |
| Alkoxyl radical RO. |
| Peroxyl radical ROO• |
| Lipidperoxides |
Table 3: Potentially toxic metabolites resulting from enzymatic degradation of ethanol in the liver.
Defining tentative molecular events of the isolated MEOS was initially not feasible, apart from the non-involvement of ADH and catalase-H2O2 [24]. Yet in 1974, we found that bovine dismutase had only a small increasing effect on the activity of the isolated MEOS, not allowing suggestions of a possible involvement of superoxide radicals [25]. In 1975, the previous observation that EtOH is an excellent scavenger of hydroxyl radicals led then to the proposal that this form of reactive oxygen species (ROS) could be a good candidate intermediate promoting microsomal EtOH oxidation [28]. Subsequently, the search for the tentative intermediate(s) promoting MEOS via its CYP 2E1 or other CYPs resulted in various reports and review articles published by several groups [5,14,39-60], including those of Lieber et al. [42-44], Coon et al. [47], Koop [46], Cederbaum et al. [47-54], Gonzalez [40,41], and Ingelman-Sundberg et al. [55-58], and a variety of these metabolic changes are considered as mechanistic cornerstones for the initiation and perpetuation of human alcoholic liver disease [60]. Although the use of different analytical approaches led occasionally to a variability of results and interpretations, the data can briefly be summarized as follows: (1) ethanol acts as a scavenger of hydroxyl radicals and undergoes oxidation to acetaldehyde during this molecular process; (2) hydroxyl radicals are generated through electron flow via the NADPH-dependent action of CYP 2E1 and other CYP isoenzymes, whereby also additional forms of ROS are produced; (3) among the other ROS intermediates of interest are superoxide radicals and H2O2, but neither of these alone can oxidize ethanol to acetaldehyde while together they may react to slowly yield hydroxyl radicals under normal conditions required for microsomal EtOH oxidation; (4) again and most importantly, H2O2 itself is incapable of promoting EtOH oxidation via the isolated MEOS that contains CYP isoenzymes but no catalase; (5) the isolated MEOS is insensitive to superoxide dismutase disregarding superoxide radicals as sole active intermediates in microsomal EtOH oxidation; (6) finally, phospholipids are required for MEOS reconstitution, serving likely as a platform for the CYP and the NADPH reductase to integrate into and thus allow for a better contact and proximity between these two enzymes to facilitate electron flow from the reductase and to reduce the ferric CYP to the ferrous state followed by binding to oxygen.
Final MEOS definition and assay conditions
Definition and specific criteria: Since half a century it is known that liver microsomes contain an enzyme system capable of oxidizing EtOH in a reaction requiring NADPH and molecular oxygen, conditions similar to those of various microsomal enzymes metabolizing drugs and other exogenous substrates [11,12]. With EtOH as substrate and known as MEOS, this enzyme system is best defined by characteristics of its isolated form, in which ADH and catalase as potential confounding contaminants have successfully been removed by column chromatography [24,25,27-29]. Instead of ADH or catalase as possible constituents of the isolated MEOS, mainstream opinion based on overwhelming evidence now is that CYP isoenzymes and the NADPH reductase are the main promoting constituents of MEOS activity (Figure 2), supported by phospholipids [5,14]. Regarding the various CYP isoenzymes involved in MEOS, agreement also exists that CYP 2E1 is the most important one generating various forms of ROS. Because EtOH is known for its radical scavenger property, it is not unexpected that hydroxyl radicals are among the most postulated intermediates promoting MEOS and thereby the metabolism of EtOH to acetaldehyde [5,14,39-60]. Since various CYP isoenzymes are constituents of MEOS, it seems plausible to define MEOS as a multi-CYP isoenzyme system.
Recommended assay conditions: For practical reasons, MEOS activity in crude liver microsomes should be determined using NADPH or a NADPH generating system contained in a specific incubation medium as described previously in detail [14]. Part of the incubation medium are among other chemicals also EDTA to prevent potential interactions by iron salts and azide to block catalase, which may contaminate crude microsomal fractions and would otherwise cause higher but unjustified metabolic rates. Clearly, azide is not part of the incubation mixture of the isolated MEOS [14] as this is free of catalase [5,14,24-29].
Hepatic microsomal CYP 2E1 specifics
Alcohol and ROS: Alcohol, ROS, and CYP 2E1 are closely interconnected with each other and determine metabolic and injurious events [5,14]. As one of the major constituents of MEOS, the isoenzyme CYP 2E1 requires as cofactor NADPH for its functions and thereby promotes the microsomal EtOH oxidation via the concomitant generation of some radicals out of the large list of intermediates as summarized under the term of ROS (Table 3) [5,14]. Initiating and keeping the CYP cycle going, the necessary electrons are provided by NADPH + H+, closely related to NADH + H+ that results from the reduction of NAD+ during ethanol metabolism via ADH [5,14]. However and prior to its metabolism through CYP, ethanol like any other substrate needs binding to CYP in its ferric state (3+), which intermittently changes to the ferrous state (2+); CYP then returns to the ferric state after completion of the oxidation and is again available for binding to the next molecule like ethanol as substrate [14]. The reactions within the CYP cycle generate ROS as toxic intermediates and by-products (Figures 2 and 3, Tables 1-3) [5,14], mostly after the uptake of two electrons and incorporation of molecular oxygen that is then incompletely split (Figure 3).

Figure 3: Cytochrome P450 cycle and substrate interaction. Ethanol as a substrate of cytochrome P450 is metabolized following several mechanistic steps involving oxygen, electrons derived from NADPH + H+, and reactive oxygen species. The original figure was published previously [30] and is reproduced with the permission of the Publisher Taylor & Francis.
ROS generated in the liver through the action of CYP 2E1 during microsomal EtOH metabolism creates microsomal oxidative stress [14] and is responsible for various effects that include metabolic alterations and membrane injury of subcellular organelles [5,14,39-60]. Confined to the endoplasmic reticulum of the liver cell and the microsomal CYP 2E1 [5], this microsomal oxidative stress has to be differentiated from the postulated mitochondrial oxidative stress due to mitochondrial CYP 2E1 [61-63].
Selected chemical substrates: A wide range of chemicals are substrates for the microsomal CYP 2E1 [14,64-85], a component of MEOS with the potential of metabolic interactions between chemicals and ethanol [5,85,86]. Among the chemicals of interest are higher aliphatic alcohols, aliphatic halogenated hydrocarbons, and many others with variable chemical structures (Table 4) [14].
Theoretically, some of these substrates could also function as potential inhibitors of MEOS activity, thereby reducing the rate of ethanol metabolism via the hepatic endoplasmic reticulum but this is poorly evaluable in humans. Vice versa and in the clinical context, ethanol can well inhibit drug metabolism [86] through interactions at the common site of CYP (Figure 4). An impaired metabolism of a substrate requires high ethanol concentrations [86]. Under these conditions, toxicity may be variable, depending on the drug used and its metabolic pathway. Conditions of interactions are different following prolonged alcohol, because liver injury is enhanced after single doses of chemicals like acetaminophen, also known as paracetamol [87,88], or carbon tetrachloride and other aliphatic halogenated hydrocarbons [89-92]. The increased toxicity results from higher metabolic rates of the used chemicals due to microsomal enzyme induction [5,14], provoked by upregulation of CYP 2E1 [5].

Figure 4: Competition of various substrates including ethanol at a common site of cytochrome P450. Ethanol and many other chemicals are substrates and metabolized by the isoenzyme CYP 2E1 but may also function as inhibitors, competing among each other and cause metabolic inhibition. The original figure was published previously [30] and is reproduced by the publisher Taylor & Francis.
Upregulation: There is yet some uncertainty about the mechanisms of the CYP 2E1 increase and related gene expression upregulation caused by chronic alcohol consumption [52,93-96]. For instance, prolonged alcohol use leads to an increased content of CYP 2E1 due to the transcription of the CYP 2E1 gene and occurs by a two– step mechanism [94] and when blood alcohol levels are high [93]. However, it has also been pointed out that increased CYP 2E1 levels are rarely accompanied by respective elevations of CYP 2E1 mRNA levels and may involve increases in gene transcription, mRNA translation, or protein stability against proteasome-mediated degradation [52]. Mechanisms of increased CYP 2E1 may be variable among the different diseases and conditions causing such increases unrelated to alcohol use [14,95-97]. Of clinical interest are increased hepatic levels of CYP 2E1 observed in patients with obesity and associated nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, diabetes mellitus, or the metabolic syndrome, as well as in fasting individuals [14]. For these conditions, acetone could be a good candidate as an endogen, natural occurring inducer [52].
As expected, much interest of CYP 2E1 upregulation and associated MEOS induction focused on molecular and mechanistic analyses related to increased metabolism of ethanol specifically via MEOS but not ADH that remained unchanged regarding its activity (Table 1) [5,14]. Through both enzymes, ethanol is degraded to acetaldehyde that is further metabolized to acetate via the NADdependent mitochondrial acetaldehyde dehydrogenase (Figure 5) [5,14,98,99].

Figure 5: Significant pathways of the hepatic alcohol and acetaldehyde metabolism. For alcohol metabolism, presented are cytosolic alcohol dehydrogenase (ADH) and the microsomal ethanol-oxidizing system (MEOS). The first toxic metabolite is acetaldehyde, which is then converted via the mitochondrial acetaldehyde dehydrogenase (ALDH) to acetate. Reproduced from a previous report [30], with permission of the Publisher Taylor & Francis.
Circulating blood plasma exosomes: In the blood plasma of patients with a history of alcohol use and of animals exposed to binge alcohol or repeated doses, extracellular vesicles containing CYP isoenzymes were detected, namely CYP 2E1, 2A6, 1A/2, and 4B in patients and CYP 2E1, 2A3, 1A/2, and 4B in animals [100]. For CYP 2E1, supporting experimental evidence was provided that blood plasma exosomes containing CYP 2E1 were released from the liver where they had been produced through ROS and oxidative stress during alcohol metabolism via CYP 2E1. Blood plasma exosomes containing CYP isoenzymes were also found in experimental animal models after an acute intoxication by acetaminophen or carbon tetrachloride [100], both chemicals are substrates for CYP 2E1 (Table 4) [14]. These innovative and encouraging data of CYP 2E1 exosomes suggest their use as diagnostic biomarkers in selected patients with acute liver disease of unknown cause or suspected alcoholic etiology, although patients in a clinical setting require an additional work up to safely exclude other, alcohol unrelated causes [5,14]. Certainly, further studies should establish the validation including sensitivity and specificity of this potential biomarker.
| Substrates | Authors |
|---|---|
| Acetaldehyde | Terelius et al., [65] |
| Acetol | Koop and Cassazza, [66] |
| Acetone | Koop and Cassazza, [66] |
| Acetaminophen | Lee et al., [67] |
| Aniline | Koop et al., [68]; Morgan et al., [69]; Nedelcheva et al., [70]; Diaz Gómez et al., [71] |
| Benzene | Nakajiama [72]; Nedelcheva et al., [70] |
| Bromobenzene | Nakajiama [72] |
| n- Butanol | Morgan et al., [73]; Lucas et al., [74] |
| Caffeine | Tassaneeyakul et al., [75] |
| Carbon tetrachloride | Johansson and Ingelman-Sundberg [76]; Nakajiama [72]; Diaz Gómez et al., [71] |
| Chloroform | Nakajiama [72]; Constan et al., [77]; Diaz Gómez et al., [71] |
| 1-Chloropropane | Nakajiama, [72] |
| Chlorzoxazone | Nedelcheva et al., [70]; Lucas et al., [74] |
| 1,1-Dichloroethane | Nakajiama [72] |
| 1,2-Dichloroethane | Nakajiama [72]; Sun et al., [78] |
| 1,1-Dichloroethylene | Nakajiama, [72] |
| cis-1,2-Dichloroethylene | Nakajiama, [72] |
| trans-1,2-Dichloroethylene | Nakajiama, [72] |
| Dichloromethane | Kim et al., [79] |
| 1,2-Dichloropropane | Yanagiba et al., [80] |
| 1,2-Dibromoethane | Wormhoudt et al., [81] |
| Diethylether | Nakajiama, [72] |
| Dimethylformamide | Nedelcheva et al., [70] |
| Cumene | Nakajiama, [72] |
| Enflurane | Thummel et al., [82] |
Table 4: Selected substrates of the hepatic microsomal CYP 2E1.
Role of MEOS in alcohol metabolism
Alcohol resorption: Following alcohol ingestion, its blood levels are higher and occur earlier if consumed preprandial with empty stomach, which enhances gastrointestinal resorption, as compared to postprandial use, which retards it resorption due to local interference with food and causes postponed maximum blood alcohol levels (Figure 6) [101].

Figure 6: Alcohol elimination under preprandial and postprandial conditions. Shown are preprandial blood alcohol concentrations due to alcohol intake prior to a meal or postprandial blood alcohol concentrations when alcohol was consumed after a meal. Phases of resorption and elimination determine the respective alcohol level in the blood. Modified from a previous article published by Salaspuro [101].
Challenge of metabolic quantification: Consensus exists that prolonged alcohol use increases hepatic MEOS activity under conditions of unchanged hepatic ADH activity [5,14]. The induced MEOS activity is due to the associated upregulation of preferentially CYP 2E1 [5] and cannot be ascribed to other enzymes such as catalase since the induction occurs also with propanol and butanol as substrates, which are not metabolized by or substrates of catalase [26-28].
In the human liver, both MEOS and ADH effectively remove most of the consumed alcohol and thereby circumvent its hazardous accumulation in the body [14]. Under normal conditions, hepatic ADH is likely responsible for a major part of the ethanol metabolism in the liver, whereas hepatic MEOS could account for 20% to 25% of the alcohol metabolism in vivo [102]. The contribution of MEOS in alcohol metabolism will considerably be increased at higher alcohol levels and following chronic alcohol use. Its high Km value for ethanol favors the role of MEOS at higher alcohol concentrations, and induction of MEOS by chronic alcohol use removes alcohol more quickly under conditions of preexisting and long lasting alcohol consumption. It has also been suggested that when corrected for microsomal losses during preparation, half to two thirds of the increase in the rate of ethanol oxidation after chronic alcohol use can be accounted for by MEOS [102]. To be on the cautious conservative side and summarizing variable results of clinical and experimental studies, MEOS may contribute >25% of overall hepatic alcohol metabolism but substantially more at high alcohol concentrations or after prolonged alcohol abuse considering mostly kinetic studies [4,14,102-116].
Various confounding variables may modify the overall alcohol metabolism, limiting the accuracy of any assessment of the contribution of MEOS. The most important potential confounding variable is the gastric ADH responsible for the first pass metabolism occurring in the gastric mucosa [117-121]. Other tentative confounders such as the existence of various hepatic microsomal CYP isoenzymes, circadian rhythms, nutritional state, and severity of the liver disease are even more difficult to evaluate quantitatively. MEOS presumably benefits from some supportive actions that may accelerate ethanol metabolism but their real impact is hardly to quantify. For instance, ethanol metabolism via ADH produces NADH + H+, its reducing equivalents can easily be transferred to NADP+ to generate NADPH + H+, the cofactor of MEOS (Figure 7). This could enhance the ethanol metabolism through both pathways, MEOS and ADH. Finally, NADH + H+ generated through ADH (Figure 7) can also be used by the NADHdependent cytochrome b5 reductase and the related cytochrome b5, accelerating thereby the ethanol metabolism via ADH. This specific reductase and cytochrome are also capable of producing ROS [122,123] to be used for ethanol oxidation.

Figure 7: Interconnected action of hepatic alcohol dehydrogenase (ADH) and the microsomal ethanol-oxidizing system (MEOS). ADH produces reducing equivalents whereas MEOS uses them, showing that both enzymes depend on each other. The original figure was published in an earlier report [30], reproduced with permission of the Publisher Taylor & Francis.
Presumably, the patient consuming alcohol will have no real benefit from clarifying the respective contributions by either ADH or MEOS. In addition, alcohol entering the liver is quantitatively metabolized to the toxic acetaldehyde, independently of the pathway used for its metabolic degradation (Figure 7). In fact, one ethanol molecule converts just to one acetaldehyde molecule but problems of increased toxicity may emerge if high amounts of acetaldehyde are generated within a short time.
Initially faced with much skepticism and heavy discussions, MEOS promoted to an interesting topic following its successful isolation and reconstitution. It is now clear that MEOS represents features similar to hepatic microsomal drug-metabolizing enzymes, functioning independently from ADH and catalase, and metabolizing ethanol to acetaldehyde via CYP isoenzymes with preference of CYP 2E1. This allows classifying MEOS as a unique, alcohol metabolizing multi-CYP isoenzyme system. Through the CYP dependent molecular cycle, a variety of toxic intermediates or radicals emerge, collectively known as ROS. Some of these actively oxidize ethanol to the toxic acetaldehyde by mechanisms involving hydroxyl radicals, events facilitated by the known radical scavenging properties of ethanol. Prolonged alcohol consumption upregulates MEOS activity and CYP 2E1 gene expression, leading to increased rates of alcohol degradation via MEOS and high amounts of toxic radicals that partially escape the scavenging properties of ethanol and cause liver injury. Based on these mechanistic steps, human alcoholic liver injury represents a molecular disease. In essence, the outlined results call for further studies to expand our knowledge on molecular specificities of MEOS in order to translate basic molecular aspects into clinical practice and therapy options.
There was no funding of this article.
The author has no conflict of interest to declare with respect to this review article.
Citation: Teschke R (2019) Biochemical Aspects of the Hepatic Microsomal Ethanol-oxidizing System (MEOS): Resolved Initial Controversies and Updated Molecular Views. Biochem Pharmacol (Los Angel) 8:267. doi: 10.35248/2167-0501.19.8.267.
Received: 05-Aug-2019 Accepted: 23-Sep-2019 Published: 30-Sep-2019 , DOI: 10.35248/2167-0501.19.8.267
Copyright: © 2019 Teschke R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.