Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2014) Volume 3, Issue 5
Bcr-Abl kinase inhibitors are very effective drugs for treatment of chronic myelogenous leukemia (CML), but treatment options are limited and relatively ineffective for patients with de-novo or acquired resistance. The K562 human erythroleukemia cell line, derived from a pleural effusion during blast crisis of a CML patient, is very useful for studying hematopoietic differentiation because it undergoes differentiation and apoptosis in response to chemicals that propagate lipid-peroxidation. 4-hydroxynonenal (4-HNE), a reactive aldehyde produced from peroxidation of polyunsaturated fatty acids, is metabolized primarily by glutathione S-transferases (GSTs). 4-HNE causes differentiation, apoptosis and necrosis in K562 cells, but cannot be used as a drug for resistant CML because of its highly toxic nature. Present studies addressed the possibility of developing an alternative targeted treatment aimed at increasing intracellular 4-HNE through inhibition of GST. Because the major GST-isoenzymes in leukemia cells are also present in normal tissues, we explored the possibility of modulating cellular 4-HNE levels by inhibiting GST isozymes with high activity towards 4-HNE. Our studies identified the presence of the GSTO1 isoenzyme in K562 cells, demonstrated its activity towards 4-HNE, and showed that its depletion causes apoptosis, necrosis and differentiation of these cells. These effects of GSTO1 depletion appear to involve RUNX1 mediated transcriptional regulation of GM-CSF. These findings offer a new target for treatment of resistant CML.
<Keywords: K562, Chronic myelogenous leukemia, GST omega GM-CSF, RUNX1, 4-HNE
4-HNE-4-hydroxynonenal; CML-chronic Myelogenous Leukemia; GM-CSF-Granulocyte Macrophage Colony Stimulating Factor; GSTO1-Glutathione S-Transferase Omega1; IL-3- Interleukin-3
Bcr-Abl kinase inhibitors are very effective and non-toxic first line therapy for chronic myeloid leukemia (CML), but de-novo as well as acquired resistance occurs in 20-30% of patients for who therapeutic options are limited [1-3]. The K562 human erythroleukemia cell line, isolated during blast crisis from a patient with CML, undergoes erythroid and myeloid differentiation and apoptosis when treated with chemicals that exert oxidative stress and propagate lipid peroxidation [4]. A reactive α, β-unsaturated aldehyde, 4-hydroxy-t-2-nonenal (4- HNE), is quantitatively the major end-product of lipid-peroxidation of eicosanoid lipids. Though 4-HNE can be metabolized further by cytochromes p450, aldehyde dehydrogenases and aldehyde reductases, the quantitatively predominant metabolic pathway for its excretion is through the glutathione S-transferase (GST)-catalyzed formation of a thioether adduct with glutathione (GSH) followed by further metabolism to a mercapturic acid by the kidney [5-8]. GSTs are the rate-limiting enzyme of the mercapturic acid pathway and several GST-isoenzymes display differential catalytic activity against 4-HNE [8-11]. Because they metabolize numerous mutagenic compounds as well as chemotherapy drugs, GSTs play a major role in carcinogenesis and cancer drug-resistance [12-15]. In human CML, they play a role in genetic susceptibility and resistance to Bcr-Abl targeted therapy [16,17]. K562 cells express multiple GST isoenzymes and modulate cell growth and apoptosis in K562 cells [11,18,19]. 4-HNE mediated effects on K562 may be due to effects on several signaling kinases (JNK, MAPK, Akt), apoptotic proteins (Bax, Bcl2) and transcription factors (Myc, Mad, GATA-1, AP-1) are known to be involved with K562 differentiation indicating that multiple simultaneous mechanisms may be operative, but the relative contribution of each or the molecular mechanisms through which 4-HNE exerts these effects are not known [20-27].
Exogenous administration of 4-HNE is not suitable as a pharmacological therapy for treatment of resistant CML because it is quite toxic. However, modulating intracellular 4-HNE by targeting enzymes that metabolize it may be a reasonable approach to consider for patients with resistant CML, and the logical choice for targeted inhibition are the GSTs. We have previously reported that K562 cells contain two quantitatively predominant cytosolic GST isoenzymes belonging to the P- and M-classes.A third low-abundance isoenzyme with pI 5.8 was also identified and shown to be immunologically cross-reactive with alpha-class GSTs (GSTA) and displayed high specific activity towards 4-HNE, but its molecular identity could not be confirmed because of its low abundance [18]. The purpose of the present studies was to reexamine the GSTs of K562 cells to identify the previously observed 4-HNE metabolizing GST and to determine whether its targeted depletion could modulate intracellular 4-HNE levels to exert differentiating or pro-apoptotic effects on K562 cells.
Reagents
4-HNE (4-hydroxy-t-2-nonenal) was obtained from Cayman Chemicals, Chicago, IL. MTT and CDNB (1-chloro-2, 4-dinitrobenzene) were procured from Sigma Chemical Co. Primary antibodies against Bax, Bcl2, JNK, pJNK, β-actin, Akt, pAkt, and cleaved PARP were purchased from Cell Signaling Technology, Inc. (Boston, MA). GSTO1 and AML1 (RUNX1) antibodies were purchased from Abcam (Cambridge, MA). Secondary antibodies goat anti-rabbit and goat anti-mouse were purchased from Cell Signaling Technology. The hGSTO1 siRNA CCUGAGUGGUUCUUUAAGAdTdT were synthesized by City of Hope Core facility, Duarte, CA. OxiSelect HNE Adduct ELISA kit was purchased from Cell Bio Labs, Inc (San Diego, CA).
Cell lines
The K562 erythroleukemia cells were obtained as frozen stock from the ATCC. The culture medium was RPMI1640 supplemented with 10% fetal bovine serum and 1% penicillin/ streptomycin; cells were grown in a 5% CO2 humidified incubator at 37°C. After 5 medium exchanges over the first two weeks, aliquots of cells were frozen. These aliquots were subsequently thawed and used for each of the studies. Mycoplasma contamination was monitored at 4 week intervals by the Mycotect kit. Doubling time was measured to determine the interval of log-phase growth and drug-treatments were performed on cells in log phase growth at a starting density of 2×104 cells /well (96-well plate). Karyotyping was performed using standard methodology in the Cytogenetics Core at City of Hope. All experiments were done with cells in log-phase growth.
Transient transfection with small interfering RNA (siRNA)
In order to knockdown the endogenous GSTO1, transient transfection was performed using Lipofectamine RNAiMAX (Invitrogen). K562 cells were centrifuged at 400xg for 5 min and resuspended in RPMI 1640 medium with 10% FBS. Then, cells were transfected with GSTO1 or scrambled control siRNA (40 nM). After 24 h, Western Blots and RT-PCR were performed to check suppression of GSTO1 protein and mRNA, respectively.
MTT assay
The MTT assay was performed using 96-well plates with 8 replicates per drug concentration and repeated on 3 separate occasions. Cell viability was assessed using trypan-blue staining and initial viable cell density was 2×104 cells per well. Treatment with 4-HNE or GSTO1 siRNA was started 12 h after inoculation of cells into wells and MTT was added to wells after 24 h exposure to either 4-HNE or GSTO1 siRNA. The assay reagents and conditions were identical to those described previously [11] and plates were read 570 nm on a Bio-Tek ELx800 ELISA plate reader.
Morphological analysis of K562 differentiation
K562 cells were inoculated into 6-well plates at 2×104 cells /mL and treated with the differentiating agent or medium alone followed by 24 h incubation at 37°C prior to cytospin on L-lysine coated slides using a CytoPro centrifuge at 400 rpm for 5 min.Wright-Giemsa stained slides were examined using an Olympus AX70 microscope with photographs at 400x magnification by a hematopathologist blinded to the treatments.
Effect of 4-HNE on apoptosis by TUNEL assay
Six-well plates were inoculated with K562 cells (2×105 /mL) in logphase growth and treated with 20 μM 4-HNE for 24 h at 37°C. Apoptosis was determined by the labeling of DNA fragments with terminal deoxynucleotidyl-transferase dUTP nick-end labeling (TUNEL) assay using Promega (Madison, WI) apoptosis detection system according to the manufacturer’s protocol.
Chromatin immunoprecipitation assay
were seeded in 100-mm plates at the density of 5×105/mL. Twenty four hours after 20 μM 4-HNE treatment, formaldehyde was added into culture media to a final concentration of 1% and the cells were incubated at room temperature for 10 min. SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.0 and PMSF) was added to the cell pellet to isolate chromatin, and the lysates were sonicated to shear DNA to an average length of 200-500 base-pairs. Input was prepared by treating aliquots of chromatin with proteinase-K, and heating at 65°C for 6 h, followed by ethanol precipitation. Pellets were re-suspended, and the resulting DNA was quantified on a NanoDrop spectrophotometer. After overnight incubation with primary antibody (AML1/RUNX1 from Abcam) at 4°C, protein G agarose beads were added to isolate immune complexes. Complexes were washed and eluted from the beads with elution buffer (1% SDS and 0.1 M NaHCO3). Cross-links were reversed by incubating at 65°C, and ChIP-DNA was purified by phenol-chloroform extraction and ethanol precipitation. PCR reactions were performed using specific primers with ChIPbound and input DNA. Results were visualized on a 1.5% agarose gel.
Western blot analyses
K562 cells were treated with differentiating agents (4-HNE and/ or GSTO1 siRNA) for 24 h followed by homogenization using a Vibra-Cell-Sonics CV188 sonicator with 3s pulses×3 at 45W. Aliquots containing 30 μg were boiled with 4x NuPAGE loading sample buffer (Life Technologies) for 5 min, resolved by NuPAGE 4-12% Bis-Tris gel and transferred to a nitrocellulose membrane. Primary antibodies were against PARP, JNK, pJNK, AKT, pAKT, Bax, Bcl2, and GSTO1 at a dilution of 1:1000. The secondary antibodies were horseradish peroxidase conjugated anti-mouse or anti-rabbit goat, and blots were developed with SuperSignal West Femto Substrate (Thermo). The loading control was β-actin (Cell Signaling Technologies, Boston, MA).
Flow cytometry
4-HNE or GSTO1 siRNA treatment was performed on log-phase cells growing at a density (5×105 cells/mL) and flow-cytometry was performed after 24 h incubation at 37°C. FITC-anti-hemoglobin antihemoglobin antibodies were used to detect intracellular hemoglobin on ethanol fixed cell and cell-surface glycophorin was detected using FITC-anti-glycophorin antibodies (anti-CD235a) on live un-fixed cells.For cell cycle distribution, cells fixed in 70% ethanol, re-suspended in 500 μl of annexin binding buffer containing 2.5 μl of RNase (stock 20 mg/ml) at 37°C for 30 min after which they were treated with 10 μl of propidium iodide (stock 1 mg/ml) solution and then incubated at room temperature for 30 min in the dark. Dual Annexin-V and propidium iodide (PI) staining was performed on live un-fixed cells. Cells were washed once with PBS and resuspended in 400 μl of cold annexin binding buffer containing 5 μl of Annexin V-fluorescein isothiocyanate for 15 min and 5 μl of 0.1 mg/ml propidium iodide for 15 min in the dark. The stained cells were analyzed using the Beckman Coulter CyAn-ADP and results were processed using ModFit Lt and Summit 4.3 Software.
Mass spectroscopic analysis of purified total cytosolic GST fraction
Total cytosolic GSTs were purified using GSH-affinity chromatography. Purity was examined by Coomassie-stained SDSPAGE and Western-blotting using antibodies to each GST-isoenzyme as described previously [28]. An aliquot containing 200 μg total purified GST protein was subjected to reduction and alkylation of cysteine using 2.5 mM dithiothreitol and 7 mM idoacetamide followed by trypsin digestion and solid phase extraction using a C18 cartridge (Supelco, Bellefonte, PA). The digested peptides were analyzed using reversephase liquid chromatography-tandem mass spectrometry analysis using a hybrid Linear ion trap (LTQ)–Fourier transform ion cyclotron resonance (FTICR, 7T) mass spectrometer (LTQFT; Thermo, San Jose, CA), which is equipped with nanospray ionization source and operated by X-Calibur [version 2.2] data acquisition software using protocols described previously [29]. Extracted ion chromatograms (areas under the corresponding chromatographic peaks) of isoform-specific doubly or triply charged tryptic peptides from the full-scan high-resolution mass spectra were then used as quantitative measures of respective GST isoenzyme expression levels.
Cloning and sequencing of hGSTO1 from K562 cells
Cells were treated with thymidine 2 mM for 24 h to synchronize them in G1/S. The homogenate was prepared by sonication of cells at 6 h after release from cell-cycle block for isolation of RNA. GSTO1 cDNA was amplified by PCR using GSTO1 specific forward primer (5’TATTTATCCATGGATGTCCGGGGAGTCA3’) and reverse primer (5’GATAAATAAGTCGACTCAGAGCCCATAGTC3’) (Biosynthesis, Lewisville, TX). Human GSTO1 cDNA was purified and cloned into T-Vector (Retrogen, Inc. San Diego, CA). The sequence of the putative GSTO1 clone was compared with the NCBI sequence (BioEdit Software, Ibis Biosciences, Carlsbad, CA) and structural and chemical similarities were compared (IBM Multiple Sequence Alignment Engine) between our pET30(a)+ vector BL21 cloned peptide and the published NCBI sequence. The hGSTO1 cDNA cleaved from T-Vector, using Nco1 and BamH1 restriction enzymes was ligated into pET30(a)+prokaryotic expression plasmid with the same restriction enzymes. The hGSTO1 cDNA from K562 in pET30(a)+vector was transfected into BL21 E.coli which were grown in bacterial culture and induced by IPTG to produce hGSTO1. GSTO1 protein was purified by GSH-affinity chromatography from the supernatant of the bacterial lysate. The identity of the purified protein was confirmed by massspectrometry.
Glutathione S-transferase activities and kinetics
For comparison of enzyme activity and kinetics of GST-isoenzymes towards 4-HNE as the substrate, we additionally cloned cDNA from GSTO1 (provided by Drs. Anneke Blackburn and Philip Board, John Curtain School of Medical Research, New South Wales, Australia), GSTO1 cloned from K562 cells, and other GST isoenzymes (GSTA1, GSTM1, and GSTP1, provided by Dr. Piotr Zimniak, UAMS, Little Rock, AR) into the pET30(a)+vector. The plasmid was expressed in E.coli-BL21 and the expressed proteins were purified by GSH-affinity chromatography. GST activity assays were performed using CDNB and 4-HNE as substrates spectrophotometrically [28].
Measurement of 4-HNE and total Lipid Hydroperoxides
Following treatment of 4-HNE or transfection with human GSTO1 siRNA or scrambled siRNA, K562 cells were harvested at 24 h and subjected to the 4-HNE adduct assay (OxiSelect HNE Adduct ELISA kit, Cell Bio Labs, Inc. San Diego, CA). The 4-HNE-adduct standard was previously prepared according to the manufacturer’s instructions and a standard curve was generated to quantify HNE-adducts.
Quantitative Real-time PCR
Total RNA was isolated from cells using the RNeasy kit (Qiagen, Valencia, CA). The cDNA was prepared using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). Real-time qPCR was performed on an ABI-7500 fast real time PCR system (Life Technologies) using Power SYBR Green master mix. The list of primer pairs used and their sequences are provided in supplemental Table 1. After initial incubation for 2 min at 50°C, the cDNA was denatured at 95°C for 10 min followed by 40 cycles of PCR (95°C for 15 s, 60°C for 60 s). The relative mRNA levels of all genes were quantified with β-actin as an internal control. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta deltaC(T)) method.
| Mr [kDA] | Km [mM] | Turnover Number Kcat [sec-1] |
CatalyticEfficiency [Kcat/Km] | |
|---|---|---|---|---|
| GSTα1 | 50 | 1.5 | 1700 | 1133 |
| GSTµ1 | 52 | 1.1 | 3145 | 2859 |
| GSTΠ1 | 47 | 0.5 | 6815 | 13630 |
| GSTO1 | 55 | 0.3 | 5133 | 17111 |
Table 1: Comparison of Kinetic Properties of GST isoenzymes towards 4-HNEa
Statistical analyses
Each experiment was repeated at least thrice to ensure reproducibility of the results. The statistical significance of differences between control and treatment groups was determined by ANOVA followed by multiple comparison tests. Differences were considered statistically significant when the p value was less than 0.05.
Cytogenetic characterization of K562 leukemia cells
The K562 cell line differentiates primarily along the erythroid pathway, though there appears to be co-expression of some megakaryocytic markers consistent with lineage infidelity. To ensure that the behavior of our model system was consistent with prior literature on the subject, we performed karyotype analysis of K562 cells using G-banding. The modal chromosome number of 66, and multiple other chromosomal abnormalities were identical or similar to those seen in previous detailed karyotypic analyses of K562 cells [30] (Figure 1). Because of the unusual appearance of chromosome 8 was at variance with the reported karyotype, we could not rule out the possibility of t (8:21) a translocation most commonly seen in AML, but it has been previously reported due to clonal evolution in end-stage, treatment resistant CML in blast crisis [31]. It encodes the AML1-ETO (aka RUNX1-ETO) fusion protein, a defective transcription factor that cannot normally enhance the transcription of the differentiation promoting factors, GM-CSF and IL3 [32]. To investigate this possibility, we performed RT-PCR using primers bridging the breakpoints, and confirmed the expression of this fusion mRNA in the cell line used for present studies. Because 4-HNE has been implicated in the regulation of AML1 expression [33], we pursued these finding with additional studies on these differentiation factors, as discussed below
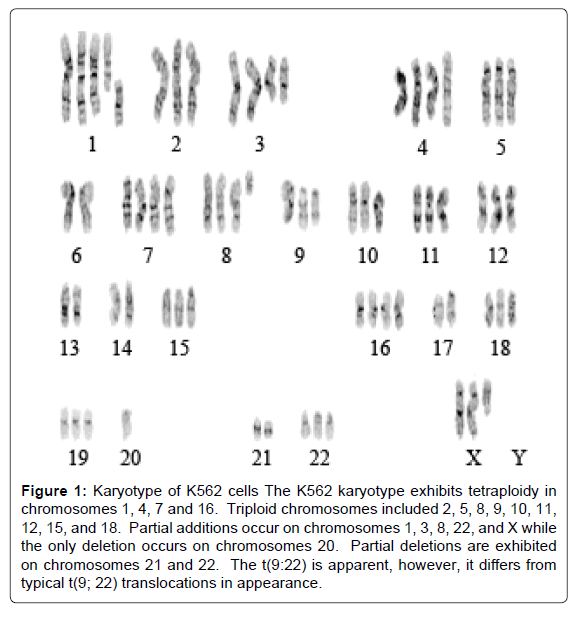
Figure 1: Karyotype of K562 cells The K562 karyotype exhibits tetraploidy in chromosomes 1, 4, 7 and 16. Triploid chromosomes included 2, 5, 8, 9, 10, 11, 12, 15, and 18. Partial additions occur on chromosomes 1, 3, 8, 22, and X while the only deletion occurs on chromosomes 20. Partial deletions are exhibited on chromosomes 21 and 22. The t(9:22) is apparent, however, it differs from typical t(9; 22) translocations in appearance.
Erythroid differentiation of K562 upon 4-HNE exposure invitro
Wright-Giemsa stain of K562 cells treated with 0.5 to 20 μM 4-HNE showed a noticeable decrease in nuclear/cytoplasmic ratio and increased perinuclear eosinophilia characteristic of cytoplasmic hemoglobin accumulation in pronormoblasts that form during erythroid differentiation (indicated by arrows). This occurred at the lowest concentration of 4-HNE used and increased in a concentration dependent manner. Nuclear fragmentation typical of necrosis was also evident at the high concentrations (Figures 2A and B). Erythroid differentiation was substantiated by flow-cytometric detection of cellsurface glycophorin expression in 4-HNE treated cells (Figure 2C). Increased accumulation of cellular 4-HNE was confirmed by ELISA assay for 4-HNE-protein adducts (Figure 3A). MTT assay showed a concentration dependent decrease in cell proliferation upon exposure to 4-HNE at concentrations between 1 and 20 μM (Figure 3B). The effector phase of apoptosis was also observed by TUNEL assay upon treatment with 20 μM 4-HNE (Figure 3C). Determination of PARPcleavage by Western-blot analysis confirmed the effector phase of apoptosis. The pro-apoptotic protein Bax was increased and the antiapoptotic protein Bcl2 was decreased. The phosphorylation of JNK, a stress-activated kinase, was increased. Activation of AKT, a survival promoting kinase, was reduced upon 4-HNE treatment (Figure 3D). At 24 h after treatment with 4-HNE, the apoptotic population quantified by flow-cytometry using Annexin-V and PI staining was increased in a concentration dependent manner. Cell cycle analysis revealed that the G1 fraction decreased and G2 fraction increased in a concentrationdependent manner with respect to 4-HNE. A dose-dependent increase in cellular debris and naked nuclei (sub-G1 population) was also observed, indicating cell lysis and necrosis (Figures 4A and B).
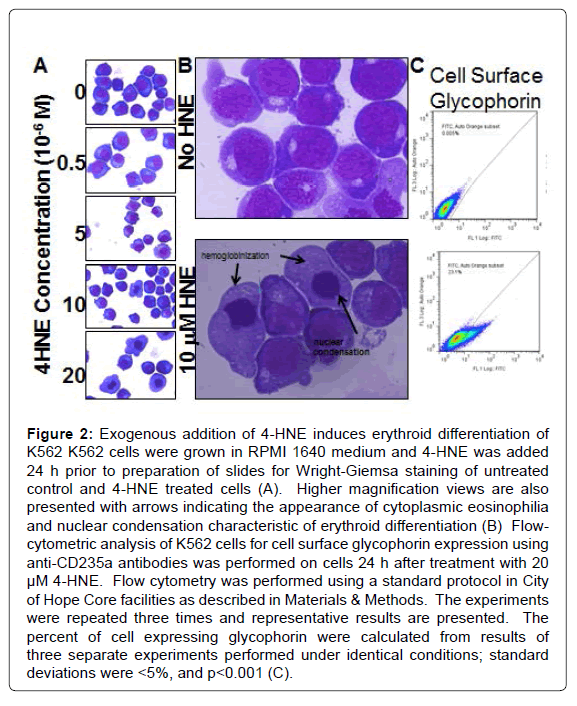
Figure 2: Exogenous addition of 4-HNE induces erythroid differentiation of K562 K562 cells were grown in RPMI 1640 medium and 4-HNE was added 24 h prior to preparation of slides for Wright-Giemsa staining of untreated control and 4-HNE treated cells (A). Higher magnification views are also presented with arrows indicating the appearance of cytoplasmic eosinophilia and nuclear condensation characteristic of erythroid differentiation (B) Flow-cytometric analysis of K562 cells for cell surface glycophorin expression using anti-CD235a antibodies was performed on cells 24 h after treatment with 20 μM 4-HNE. Flow cytometry was performed using a standard protocol in City of Hope Core facilities as described in Materials & Methods. The experiments were repeated three times and representative results are presented. The percent of cell expressing glycophorin were calculated from results of three separate experiments performed under identical conditions; standard deviations were <5%, and p<0.001 (C).
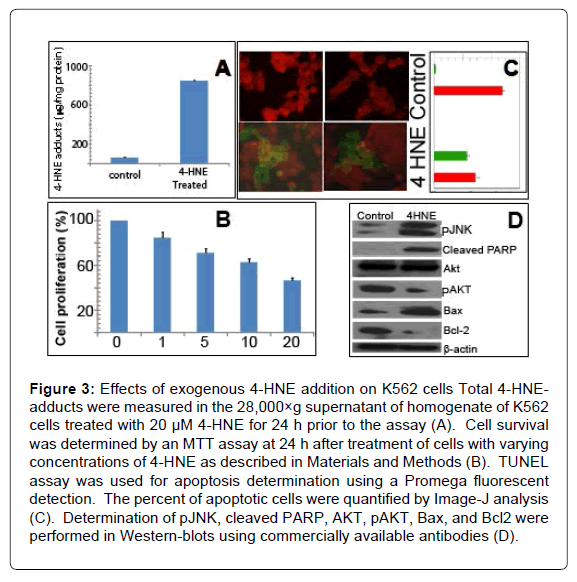
Figure 3: Effects of exogenous 4-HNE addition on K562 cells Total 4-HNE-adducts were measured in the 28,000×g supernatant of homogenate of K562 cells treated with 20 μM 4-HNE for 24 h prior to the assay (A). Cell survival was determined by an MTT assay at 24 h after treatment of cells with varying concentrations of 4-HNE as described in Materials and Methods (B). TUNEL assay was used for apoptosis determination using a Promega fluorescent detection. The percent of apoptotic cells were quantified by Image-J analysis (C). Determination of pJNK, cleaved PARP, AKT, pAKT, Bax, and Bcl2 were performed in Western-blots using commercially available antibodies (D).
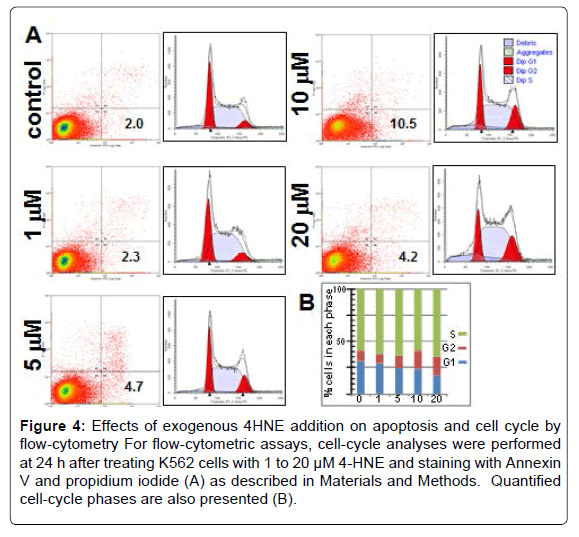
Figure 4: Effects of exogenous 4HNE addition on apoptosis and cell cycle by flow-cytometry For flow-cytometric assays, cell-cycle analyses were performed at 24 h after treating K562 cells with 1 to 20 μM 4-HNE and staining with Annexin V and propidium iodide (A) as described in Materials and Methods. Quantified cell-cycle phases are also presented (B).
GST isoenzymes of K562 cells
Total GSTs were purified from K562 cells using conventional GSHaffinity chromatography and Coomassie-stained SDS-PAGE showed the presence of three protein bands at 23, 26 and 27.5 kDA in this fraction. Western-blot analyses using isoenzyme-specific antibodies confirmed our previous findings, identifying that the 23 kDA band as GSTP and the 26 kDA band as GSTM. The 27.5 kDA was cross-reactive with antibodies towards GSTA, but was not recognized by antibodies against GSTA4, an alpha-class human GST isoenzyme with high activity towards 4-HNE. Sequencing of peptides in the total purified GST fraction by MS-MS confirmed the presence of GSTP and GSTM, and showed the presence of GSTO1. GSTO1 specific primers were used to clone full-length GSTO1 from K562 cells and its identity was confirmed by complete sequencing of the cDNA. A novel substitution mutation A140V was identified by DNA sequencing. The hGSTO1 cDNA was cloned into the pET30(a)+ prokaryotic expression plasmid. The GSTO1 clone from K562, the wild-type clones of GSTO1, GSTA1, GSTM1 and GSTP1 were expressed in E.coli BL21 and purified. In kinetic studies, the catalytic efficiency (Kcat/Km) of the GSTO isoenzymes towards 4-HNE was greater than the other GST isoenzymes, and the A140V mutation did not affect the activity (Table 1).
Apoptosis and erythroid differentiation of K562 upon depletion of GSTO1
Transfection of GSTO1 siRNA (67 ± 11% transfection efficiency) significantly reduced GSTO1 mRNA (52 ± 7%, p<0.04) as measured by qRT-PCR. Western blot analysis confirmed the concomitant depletion of GSTO1 protein (Figure 5A). GSTP and GSTM mRNA quantified by qRT-PCR were unaffected by GSTO1 siRNA treatment. The expression of GM-CSF and IL3 was slightly, but significantly increased (p<0.05); in contrast, mRNA for the AML1-ETO fusion protein was remarkably decreased (Figures 5B and 6F). GSTO1 depletion caused erythroid differentiation in Wright-Giemsa stains and apoptosis in TUNEL assays. The degree of differentiation assessed by appearance of erythroid pronormoblasts (indicated by arrows) was less than seen with exogenously added 4-HNE (Figures 5C and 5D). Treatment of GSTO1 depleted cells with 4-HNE increased differentiation at 10 μM 4HNE, whereas apoptosis and necrosis predominated at 20 μM (Figures 5E and 5G) as compared with that seen with 4-HNE alone see Figure 2. The apoptotic population by TUNEL assay in cells treated with 20 μM 4-HNE without GSTO1 depletion (44 ± 6%, Figure 3C) was significantly lower than those with GSTO1 depletion (73 ± 9%, with 854 ± 68 cells counted, n=3, p<0.01). GSTO1 depletion also caused the appearance of cytoplasmic hemoglobin and cell-surface glycophorin by flowcytometry (Figure 6A). MTT assay revealed a 35 ± 7% decrease in cell proliferation upon partial depletion of GSTO1 (Figure 6B). Depletion of GSTO1 was accompanied by a 1.7 fold (p<0.01) increase in cellular 4-HNE-adducts (Figure 6C). Western-blot analysis of PARP-cleavage confirmed apoptosis caused by GSTO1 depletion; decreased pAKT, and increased Bax were also apparent (Figure 6D). The apoptotic and necrotic effects of GSTO1 depletion alone were less pronounced than with 20 μM HNE alone, consistent with a greater increase in cellular 4-HNE by the latter treatment.
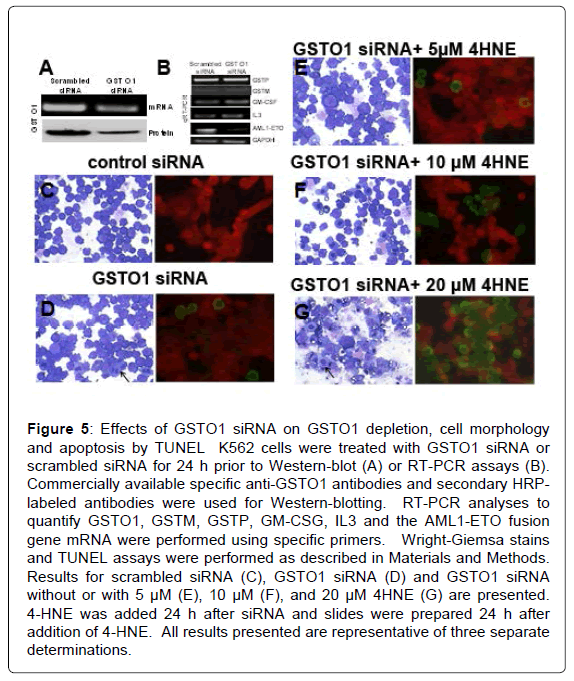
Figure 5: Effects of GSTO1 siRNA on GSTO1 depletion, cell morphology and apoptosis by TUNEL K562 cells were treated with GSTO1 siRNA or scrambled siRNA for 24 h prior to Western-blot (A) or RT-PCR assays (B). Commercially available specific anti-GSTO1 antibodies and secondary HRPlabeled antibodies were used for Western-blotting. RT-PCR analyses to quantify GSTO1, GSTM, GSTP, GM-CSG, IL3 and the AML1-ETO fusion gene mRNA were performed using specific primers. Wright-Giemsa stains and TUNEL assays were performed as described in Materials and Methods. Results for scrambled siRNA (C), GSTO1 siRNA (D) and GSTO1 siRNA without or with 5 μM (E), 10 μM (F), and 20 μM 4HNE (G) are presented. 4-HNE was added 24 h after siRNA and slides were prepared 24 h after addition of 4-HNE. All results presented are representative of three separate determinations.
Effect of 4-HNE on expression of AML1 target genes
The remarkable effects of GSTO1 depletion on the AML1-ETO fusion mRNA led us to examine whether 4-HNE treatment affected the binding of this factor to regulatory elements of GM-CSF and IL3. 4-HNE treatment increased GM-CSF protein (p<0.01) (Figure 6E); IL-3 protein was also increased slightly at the protein level, though the effect was not statistically significant (p<0.1, data not presented).A qRT-PCR analysis revealed that GM-CSF, IL3, and glycophorin (CD- 235a) mRNA were increased significantly upon depletion of GSTO1 by siRNA (n=3, p<0.01) (Figure 6F). Since the AML1 transcriptionally regulates both GM-CSF and IL3, a CHiP assay was performed using anti-AML1 antibody for pull-down. Treatment with 4-HNE increased AML-1 (or AML1-ETO) binding to the upstream regulatory sequences of GM-CSF and IL3, though the effect on GM-CSF was greater.Because the precipitating antibodies were to AML1, it cannot be ascertained whether AML1 or the fusion protein were bound to the transcriptional regulatory elements. Control IgG did not pull down either GM-CSF or IL3 regulatory elements (Figure 6G).
Results of our studies demonstrated 4-HNE causes concentration dependent differentiation, apoptosis and necrosis of K562 cells. Our results demonstrated for the first time that these cells express a mutant GSTO1 at aa 140 (A140V), a site at which a different mutation (A140D) is associated with increased risk for pediatric acute lymphoblastic leukemia [60]. GSTO1 displayed high catalytic efficiency towards 4-HNE, unaffected by the mutation. The present results support prior studies showing that genetic instability can give rise to the t(8;21) translocation in cultured K562 cells in a manner similar to that seen in-vivo. Targeted depletion of GSTO1 increased cellular 4-HNE and caused differentiation, apoptosis and necrosis. Treatment with 4-HNE resulted in up-regulation of GM-CSF mRNA and protein, accompanied by down-regulation of the transcriptional repressor, AML1-ETO1. These studies provide new information to develop novel treatment strategies for treatment of resistant CML by targeting the mercapturic acid pathway, and indicate the need for more detailed examination of mechanisms of transcriptional regulation by 4-HNE.
Several lines of evidence supported the therapeutically relevant effects of 4-HNE. Differentiation was evident from cytological studies showing widely accepted cellular changes indicative of hematopoietic differentiation, from cytometric studies of glycophorin expression. These results strengthen our previous findings of increased hemoglobinization of K562 cells upon 4-HNE exposure, and the protective effect of transfection of cells with another 4-HNE metabolizing enzyme, GSTA4 [11]. 4-HNE induced apoptosis was also demonstrated through complementary approaches including cytology, MTT assay, TUNEL assay, flow-cytometry of PI/annexin-V stained cells, changes in cell cycling and accumulation of the sub-G1 population. Discrepancy in the extent of apoptosis between the TUNEL and flow-cytometric assays is characteristic of K562 and other cell types, and is dependent on drug-concentration and time of exposure [34,35]. The observed G2 arrest with 4-HNE in a manner similar to alkylating agent drugs suggests that the effects of 4-HNE may be due to DNA-alkylation [36]. The greater effect on apoptosis of exogenous 4HNE as compared with GSTO1 depletion may be due to an inherent difference in the two techniques for increasing cellular 4-HNE. Because the volume of culture medium is ‘infinitely’ larger than cell volume, addition of 4-HNE to the medium essentially provides an unlimited supply of 4HNE resulting in greater overall exposure that can cause extensive protein and DNA cross-linking. In addition, the subcellular distribution of endogenously generated 4-HNE is dependent on differential production and metabolism of 4HNE in sub-cellular compartments; this cannot be replicated by exogenous 4-HNE inherently different biological effects are expected as seen in previous studies by others [37,38].
We considered the possibility that off-target effects of GSTO1 depletion by siRNA were responsible for the observed effects. Potential indirect effects unrelated to its GST activity, such as perhaps signaling or transcriptional changes that increase the oxidation of polyunsaturated fatty acids cannot be completely ruled out. A non-specific effect of siRNA is unlikely since the scrambled siRNA did not affect cellular 4-HNE, differentiation or apoptosis. Off target effects related to depletion of other GST isoenzymes are also unlikely because GSTO1 siRNA did not deplete GSTP or GSTM. Because there is no significant structural or sequence homology of GSTO1 with the only other 4-HNE metabolizing enzymes (cytochromes p450, aldehyde /aldose reductases or aldehyde dehydrogenases), it is very unlikely that increased cellular 4-HNE was due to a direct effect of the siRNA on these enzymes.The high catalytic efficiency of GSTO1 towards 4-HNE, the rise in cellular 4-HNE upon GSTO1 depletion, and the synergy between GSTO1 and exogenous 4-HNE also argue against the possibility that off-target effects are responsible for the observed apoptosis or differentiation.
4-HNE induces granulocytic maturation in the HL60 AML cell line known to express AML1. 4-HNE causes growth inhibition, apoptosis and necrosis in the CML-origin K562 erythroleukemia and increases surrogate markers of erythroid differentiation [20], but convincing morphological evidence for hematopoietic differentiation of K562 cells has not previously been presented. Unlike the differentiating effect in K562 cells, 4-HNE inhibits erythroid differentiation in non-malignant hematopoietic cells [39]. This indicates cell-dependent differential effect of 4-HNE and suggest a fundamentally opposite effect between normal and malignant erythroid cells, and could translate into a greater therapeutic index.
Because 4-HNE is not sufficiently reactive to directly initiate lipidperoxidation, its actions are likely mediated through its alkylating activity towards signaling proteins and DNA. α,β-alkenal compounds such as 4-HNE directly bind DNA and modulate AP-1 [40,41], which in turn regulates the expression of GST, AML-1, IL-3, GMCSF and β-hemoglobin. AP-1 and AML1 cooperate in hematopoietic differentiation by regulating the expression GM-CSF [24,41-46]. Our results indicated that 4-HNE regulates the expression of GM-CSF and IL3, associated with a remarkable down-regulation of the AML1-ETO fusion protein, through unknown mechanisms that require further investigation. Translocation involving AML1 are frequently seen in AML, a leukemia in which 4-HNE analogs have anticancer activity [47-49]. Though AML1 translocations are not seen in de-novo CML they are observed in drug-resistant CML during blast crisis. Thus GSTO1 targeting may be clinically applicable in drug-resistant CML for which there are few good options of therapy. Indeed, given the recently described role of GSTO1 in other malignancies, exploration of the implications of our findings in other cancer would also be of interest [48,49].
The potential therapeutic implications of our findings need to be considered in the wider context of targeting the mercapturic acid pathway for therapy of drug-resistant malignancy. The activity of the mercapturic acid pathway is frequently increased in treatment-resistant malignancy and this pathway plays a key role in the metabolism and excretion of 4-HNE and other pro-apoptotic electrophilic metabolites of lipid peroxidation. GST enzymes, the first committed step in the mercapturic acid pathway, catalyze the formation of thioether adducts of GSH with electrophiles such as 4-HNE. These GSH-electrophile adducts are removed from cells through energy dependent transport by membrane transporters such as RLIP76/RALBP1 prior to further metabolism to mercapturic acids by the kidney. A central role of RLIP76 in regulating cellular 4-HNE is clearly evident form studies that show marked increase in 4-HNE levels in tissues of RLIP76 knockout mice. Regression of multiple types of cancer with accompanying increase in cellular 4-HNE and lack of significant normal cell toxicity upon blocking RLIP76 strongly supports a key anti-apoptotic role of the mercapturic acid pathway in malignant cells and indicates that cancer cell-specific apoptosis is an inherent property of 4-HNE [50,51]. Its relevance in CML has also been demonstrated in studies showing apoptosis of K562 cells upon its blockade [52]. It is thus possible that combined targeting of GSTO1 and RLIP76 could be an even more effective therapy. Further investigations are needed to directly test this speculation.
In summary, our studies have demonstrated the expression of GSTO1 in a CML-derived cell line. Targeted depletion of GSTO1 increases cellular 4-HNE levels and causes differentiation, apoptosis and necrosis of K562 cells in a manner similar to that observed with exogenous treatment with 4-HNE. Modulation of intracellular 4-HNE by specific depletion of 4-HNE could be of clinical relevance in treatment of drug-resistant CML.
These studies were supported by NIH grant CA77495 to SA. The authors wish to thank Dr. Xiangle Sun (University of North Texas Health Science Center, Fort Worth, TX, Microscopy Facility) as well as Dr. Brian Armstrong, Ms. Mariko Lee, and Professor Ivan Todorov for help in microscopy (City of Hope). The authors also wish to thank the City of Hope Cytogenetics Core for karyotype assistance and the Analytical Cytometry Core for help in performing flow-cytometry and the Mass Spectrometry Facility at University of North Texas Health Science Center, Fort Worth, TX. KL also wishes to acknowledge her parents Beverly and James Leake, who courageously battled cancer and inspired her, and especially her husband Mr. Robert Bair, Jr., who has always been supportive.
KL was the principal contributor in formulating the hypothesis, search and interpretation of literature, experimental protocol design and execution, and manuscript preparation. She was aided in performance of experiments by JS and SSS. SA is the senior author who personally supervised and advised KL in design and conduct of experiments, data analysis and manuscript preparation. KL and SA were aided by SSS in data collection, analysis, and manuscript preparation.