Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2013) Volume 2, Issue 1
Most countries have adopted the good clinical practice (GCP) principles as laws and regulations for approving clinical trials which are conducted to test new methods to prevent, detect, and treat disease or drug development. Most regulatory agencies such as the United State Food and Drug Administration (FDA) indicate that only adequate and well-controlled clinical trials can provide substantial evidence of safety and effectiveness of the test treatment under investigation. FDA indicates that “substantial evidence” can only be provided through “adequate and wellcontrolled” clinical investigations which include valid study design and appropriate statistical methods for data
analysis. In this article, we are going to highlight the key points in design and analysis of clinical trials. We will also review the current research and point out the issues for future research.
Few commonly employed study designs and its concepts in clinical trials will be described. In addition, statistical methods for data analysis on efficacy and safety are reviewed. To maintain the integrity and quality of the intended trial, Good Clinical Practice (GCP) and independent Data Monitoring Committee (DMC) are also discussed. Recent development such as biomarker development for target clinical trial, translational research/medicine, and traditional Chinese medicine are also provided.
Keywords: Clinical trial designs, Efficacy, Safety, GCP, DMC
In biomedical research, clinical trial is necessarily conducted to test new methods to prevent, detect, and treat disease or drug development under investigation is safe and efficacious. Since clinical trials are usually involving humans, they can only conducted after satisfactory information has been gathered and been approved by the independent ethics committee or the health authority in the country. The conduct of clinical trial is to collect quality and representative data for a valid and unbiased assessment of the test treatment under investigation. Most regulatory agencies such as the United State Food and Drug Administration (FDA) indicate that only adequate and wellcontrolled clinical trials can provide substantial evidence of safety and effectiveness of the test treatment under investigation [1-8].
Most countries have adopted the Good Clinical Practice (GCP) principles as laws and regulations [9-14]. These regulations are enforceable but an alternative approach may be used if such approach satisfies the requirements of the applicable statute and regulations. The effectiveness requirement for drug approval was added to the Federal Food, Drug, and Cosmetic Act (FD and C Act) in 1962. After two years of public hearings on this issue, the US Congress adopted the amendments and defined “substantial evidence” as “adequate and wellcontrolled investigations, including clinical investigations.” (21 CFR- 314) [13]. An adequate and well-controlled study has the following characteristics that (1) the study objectives are clearly stated; (2) use a design that permits a valid comparison with a control to provide a quantitative assessment of drug effect; (3) the method of selection of subjects provides adequate assurance that they have the disease or condition being studied, or evidence of susceptibility and exposure to the condition against which prophylaxis is directed; (4) the method of assigning patients to each groups minimizes bias and is intended to assure comparability of the groups with respect to pertinent variables such as age, sex, severity of disease, duration of disease, and use of drugs or therapy other than the test drug; (5) adequate measures such as blinding are taken to minimize bias on the part of the subjects, observers, and analysts of the data; (6) the methods of assessment of subjects’ response are well-defined and reliable; (7) valid analysis for an unbiased and reliable assessment of the effect of the drug.
Relevant guidance in clinical trial are constructed in many countries and many researches articles about trial designs and analyses methods to variety of experimental purposes are published in the past several decades. In this article, we are not going to introduce each designs and analyses method in detail but highlight the key points in commonly used designs and analyses methods of clinical trials. We will also review the current research and point out the issues for future research.
In what follows, few commonly considered clinical trial designs and statistical methods for data analysis are described.
Chow and Liu [15] indicated that two important aspects for the conduct of a clinical trial are “How to choose an appropriate study design?” and “How to analyze the collected clinical data using valid statistical methods?” A well-organized study protocol is often developed to address these questions The FDA indicates that a wellorganized protocol should include study objective(s), study design, patient selection criteria, dosing schedules, statistical methods, and other medical related details.
In practice, different trial designs may be employed for achieving different study objectives. For example, a dose finding (escalation) design is often employed for identifying the Minimum Effective Dose (MED) and/or Maximum Tolerable Dose (MTD) in early clinical development, which is often considered the optimal dose for subsequent clinical studies conducted at later phase of clinical development. Since an inappropriate study design may not capture the right or correct data for addressing the study objectives of the intended clinical trial. Some commonly used clinical trial designs are briefly described below.
Crossover design versus parallel design
The most commonly used clinical trial design is probably a parallel design (Figure 1), which usually consists of two treatment groups. Qualified subjects are randomly assigned to receive each of the two treatments at either a 1:1 ratio or an unequal ratio of treatment assignment. Relative to the parallel design, a standard two-sequence, two-period crossover design (Figure 2) includes two sequences of treatments. Qualified subjects are randomly assigned to receive each of the two sequences of treatments. For example, subjects who are assigned to receive the first sequence of treatments will receive treatment A first and then crossover to receive treatment B after a sufficient length of washout.
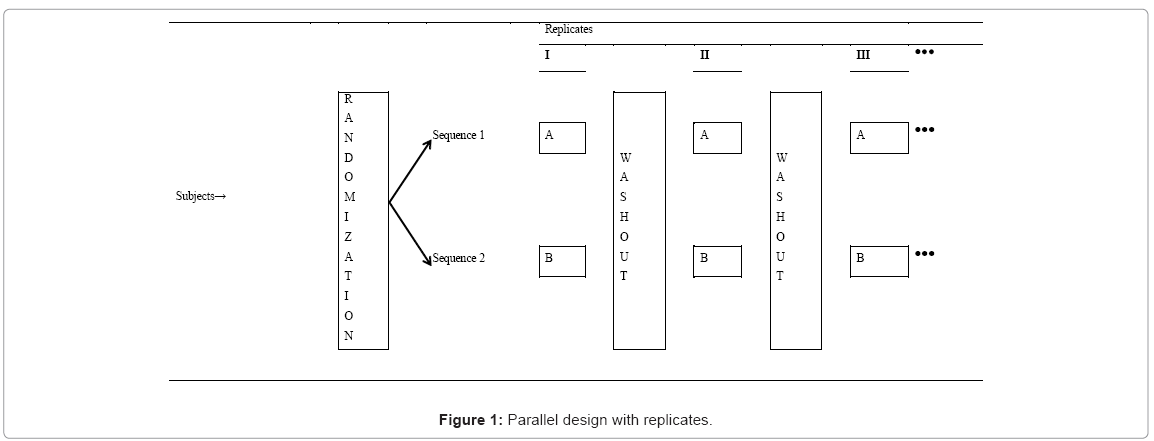
Figure 1: Parallel design with replicates.
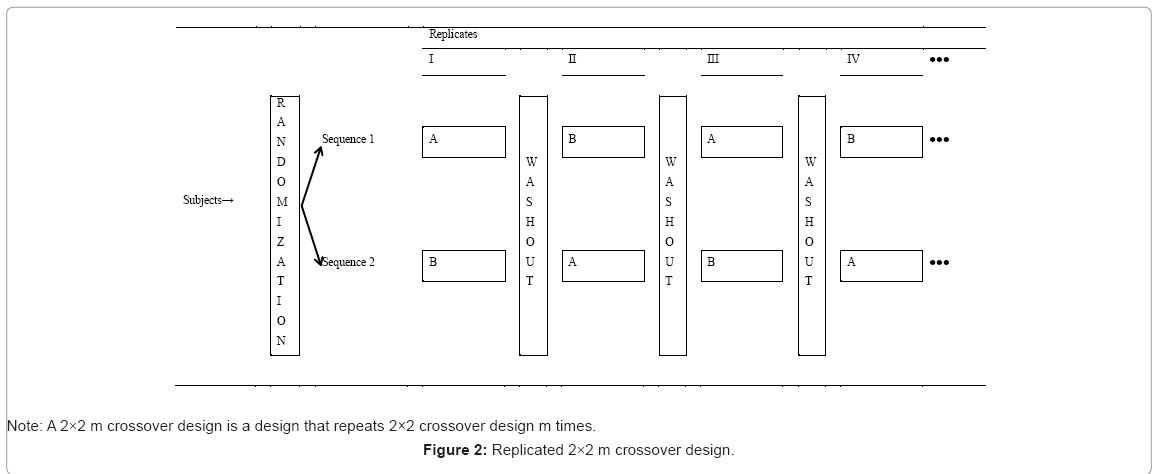
Figure 2: Replicated 2×2 m crossover design.
Parallel design is simple and easy to implement, which is applicable to acute conditions (e.g., infection or myocardial infarction). Analysis is less complicated, and interpretation of the results is straightforward. A parallel group design is probably the most commonly used design in phases II and III of clinical trials. However, it usually requires more patients than other comparative designs. Unlike a parallel design, a, crossover design can reduce the influence of confounding covariates because experiment subjects serve their own control. It allows a withinpatient comparison between treatments and removes inter-patient variability from the comparison between treatments. Also, an optimal crossover designs are statistically efficient which provides the best unbiased estimates for the differences between treatments, so require fewer subjects than parallel study.
Dose finding design
Dose finding design is usually conducted to determine which dose works best or which dose is least harmful. In phase I or early phase II clinical trial, a dose-finding design can help in determining the optimal biological dose (OBD) of a drug based on the MTD of certain toxicity rate, efficacy or low risk of side effects. However in clinical trials, it is unethical to assign patients to high doses until lower ones have been explored. In addition, we do not wish to put too many subjects at low, ineffective doses. The design includes a placebo group of subjects, and a few groups that receive different active doses of the test drug. Based on these principals, commonly seen dose finding (escalation) designs in cancer research are traditional 3+3 dose escalation trial design and the design utilizing so-called Continuation of Re-assessment Method (CRM) [16].
In clinical trials, the 3+3 trial design is often used without any scientific justification. Chow and Liu [15] suggest that the probability for achieving the MTD be used to justify the selected design for a fixed selected sample size. In addition, relative advantages and limitations of the two methods should be carefully evaluated for selecting an appropriate study design for dose finding. For example, 3+3 tends to under-estimate the MTD while CRM has smaller number of subjects exposed to DLT (Dose Limiting Toxicity) prior to achieving the MTD. The CRM generally has higher probability of reaching MTD with less number of subjects. The CRM can be further improved in conjunction with Bayesian approach when updating the dose-toxicity model.
Group sequential design
Group sequential design is a design that allows for prematurely stopping a trial due to safety, futility/efficacy or both with options of additional adaptations based on results of interim analysis. Various stopping boundaries based on different boundary functions for controlling an overall type I error rate are available. For Long-term and life-threatening clinical trials, FDA recommends group sequential design [17] because it is to gather information, monitor data in the process that the experiment can be stop or issued prior to the completion of all data collection in ethical considerations. Based on the analysis results in period, vendors can decide whether to adjust the design.
Adaptive design
The purpose of adaptive design methods in clinical trials is to give the investigator the flexibility for identifying any signals or trends (preferably best or optimal clinical benefit) of the test treatment under investigation without undermining the validity and integrity of the intended study. FDA defined that adaptive design is as a study including a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of data (usually interim data) from subjects in the study. Based on the type of adaptations (prospective, concurrent, and retrospective adaptations), Chow and Chang [18] classified adaptive designs into the following categories: (1) an adaptive randomization design, (2) a group sequential design, (3) a flexible sample size re-estimation (SSR) design or an N-adjustable design, (4) a drop-the-loser (or pick-the-winner) design, (5) an adaptive dose finding design, (6) a biomarker-adaptive design, (7) an adaptive treatment-switching design, (8) an adaptivehypothesis de-sign, (9) an adaptive seamless (e.g., a two-stage phase I/II or phase II/III) trial design, and (10) a multiple adaptive design, which is any combinations of the above adaptive designs.
Enrichment design
Some therapeutic agents are likely to be effective in a specific population of patients who may have an underlying disorder that is responsive to the manipulation of dose levels of the same agent or several different agents. Temple [19] indicates that there are basically three kinds of enrichment: noise reduction, prognostic and predictive. Enrichment won’t save a drug that doesn’t work, but it will help find one that does. An enrichment design should enhance signals of effectiveness. Instead of an unselected group of patients, enrichment design is of interest to identify the patients in whom the test agent is likely to be beneficial in the early phase of the trial. The patients with drug efficacy identified at the enrichment phase are then randomized to receive either the efficacious dose of the test agent or the matching placebo. The concept of enrichment design is illustrated in three clinical trials in the areas of Alzheimer’s disease and arrhythmia.
In clinical trials, statistical considerations of the analytic process include the eligibility criteria, randomization, treatment regimens (e.g., dose level, schedule, and treatment duration), sample size estimation (justification and/or re-estimation), planned schedule of patient evaluations for data collection (e.g., number of intermediate time points, timing of last patient observation and duration of patient study participation), primary study endpoint (e.g., which of several types of outcome assessments, which time point of assessment, use of a unitary versus composite endpoint or the components included in a composite endpoint, secondary endpoints, analytic methods to evaluate the endpoints (e.g., covariates of final analysis, statistical methodology, Type I error control), the criteria for efficacy and safety assessment, possible interim analysis and data monitoring, and statistical and clinical inference. By defining response variables (or clinical endpoints), efficacy and safety can be clearly indicated.
Safety
FDA requires a minimum of 30 subjects be studied at the highest dose of the treatment must be included in the safety analysis. Chow and Liu [15] indicated that the primary safety variable is the incidence of Adverse Event (AE), which is defined as any illness, sign, or symptom that has appeared or worsened during the course of the clinical study regardless of causal relationship to the medicine under study. Although present AE data by tables and graphics provide useful information for the safety evaluation of a study drug, they do not provide any statistical inference for the safety assessment.
For rare adverse events the primary interest is to estimate the occurrence. For common adverse events, it is also of interest to make comparisons between subgroups. Basically the analysis of AE depends on the type of data, which can be classified as nominal (binary) or ordinal, counts or rates, or time to occurrence which are usually analyzed by Fisher’s exact test or the Mantel-Haenszel test, logistic regression, and survival analysis. For other types of adverse events data such as absorbing events and recurring events with and/or without duration, statistical methods including the Kaplan-Meier and the Cox proportional hazards models can be directly applied.
Compare to large sample approach of Mantel-Haenszel test, Fisher’s exact test is often used when the sample size is small. And the Cochran-Mantel-Haenszel (CMH) test is useful when take the study site (or center) into consideration. In practice, one may also interested in investigating whether the number of subjects at which pre- and post-treatment disagreed was distributed by them in a similar manner among other categories. In testing categorical shift, the Stuart-Maxwell test is often considered.
Efficacy
Efficacy endpoints may be direct measures of clinical benefit (e.g., improved survival or alleviation of symptoms) or they may be laboratory measurements or physical signs expected to correlate meaningfully with clinical benefit. By identifying the appropriated primary efficacy variable, the efficacy can be assessed both in vivo and in vitro.
For efficacy evaluation of the drug product under investigation, formal statistical inferential procedures are usually performed to establish the benefit of the treatment based on the efficacy data. However, many clinical and statistical issues may be raised during the analysis of efficacy data. These issues need to be addressed before a fair and unbiased assessment of efficacy can be reached. The FDA guidelines on the format and content for the full integrated clinical and statistical report and the ICH guidelines [20-29] on the structure and contents of clinical study reports require the following issues be addressed in the final reports: (1) baseline comparability, (2) analyses of the intentionto- treat dataset versus evaluable dataset, (3) adjustments of covariates, (4) multicenter trials, (5) subgroups analysis, (6) multiple endpoints, (7) interim analysis and data monitoring, (8) active control studies, and (9) handling of dropouts or missing data.
For evaluating the efficacy of several test drugs for the same indication compared to a placebo control, the Analysis Of Variance (ANOVA) method can be applied to compare several population means. If it is believed that the endpoints are usually linearly related to the baseline values, an adjusted analysis of variance should be considered to account for the baseline values. This adjusted analysis of variance is called Analysis Of Covariance (ANCOVA). For the analysis of censored data, the survival function and the use of the Kaplan- Meier’s method, Wilcoxon rank sum statistic for the censored data will be used to evaluate the efficacy.
Good Clinical Practices (GCPs) considered by the International Conference on Harmonization (ICH) is a set of international ethical and scientific quality standards for designing, conducting, recording, and reporting trials that involve human subjects. In most countries, governments usually transpose GCP into regulations for clinical trials involving human subjects. Compliance with GCP assures that the human rights, safety and efficacy are credible in the clinical trial.
GCP guidelines include (1) standards on “how clinical trials should be conducted? (2) definition of the roles and responsibilities of clinical trial sponsors, clinical research investigators, and monitors. The table 1 presents the list of ICH guidelines related to GCP.
| Codes | Topic | Finalized in | Description |
|---|---|---|---|
| E3 | Structure and Content of Clinical Study Reports | November 1995 | This document describes the format and content of a study report that will be acceptable in all three ICH regions. It consists of a core report suitable for all submissions and appendices that need to be available but will not be submitted in all cases |
| E5 | Ethnic Factors in the Acceptability of Foreign Clinical Data | February 1998 | This document addresses the intrinsic characteristics of the drug recipient and extrinsic characteristics associated with environment and culture that could affect the results of clinical studies carried out in regions and describes the concept of the "bridging study" that a new region may request to determine whether data from another region are applicable to its population. |
| E6 | Good Clinical Practice | May 1996 | This Good Clinical Practices document describes the responsibilities and expectations of all participants in the conduct of clinical trials, including investigators, monitors, sponsors and IRBs. GCPs cover aspects of monitoring, reporting and archiving of clinical trials and incorporating addenda on the Essential Documents and on the Investigator's Brochure which had been agreed earlier through the ICH process. |
| E7 | Studies in Support of Special Populations: Geriatrics | June 1993 | This document provides recommendations on the special considerations which apply in the design and conduct of clinical trials of medicines that are likely to have significant use in the elderly. |
| E10 | Choice of Control Group and Related Issues in Clinical Trials | July 2000 | This document addresses the choice of control groups in clinical trials considering the ethical and inferential properties and limitations of different kinds of control groups. It points out the assay sensitivity problem in active control equivalence / non-inferiority trials that limit the usefulness of trial design in many circumstances. |
| Q9 | Quality Risk Management | November 2005 | This Guideline provides principles and examples of tools of quality risk management that can be applied to all aspects of pharmaceutical quality including development, manufacturing, distribution, and the inspection and submission/review processes throughout the lifecycle of drug substances and drug (medicinal) products, biological and biotechnological products, including the use of raw materials, solvents, excipients, packaging and labeling materials. |
| GL9 | Good Clinical Practice | June 2000 | The objective of this document is to provide guidance on the design and conduct of all clinical studies of veterinary products in the target species |
*Resource: International Conference on Harmonization Tripartite Guidelines
Table 1: Summary of Selected ICH guidelines related to GCP.
ICH guidelines [20-29] provide a unified standard for the European Union, Japan, and the United States to facilitate the mutual acceptance of clinical data by the regulatory authorities in those jurisdictions.
To ensure the ethic and quality data, FDA regulates scientific studies that are designed to develop evidence to support the safety and effectiveness of investigational drugs (both human and animal), biological products, and medical devices. The following resources are provided in the Code of Federal Regulations (CFR) [10-13] to assist investigators, sponsors, and contract research organizations who conduct clinical studies on investigational new drugs comply with the U.S. law and regulations covering GCP (Table 2).
| Regulations | Title | Description |
|---|---|---|
| Part 50 | Protection of Human Subjects | The regulations clarify existing FDA requirements governing informed consent and provide protection of the rights and welfare of human subjects involved in research activities that fall within FDA's jurisdiction. |
| Part 56 | Institutional Review Boards | This part contains the general standards for the composition, operation, and responsibility of an Institutional Review Board (IRB) that reviews clinical investigations regulated by the FDA. Compliance with this part is intended to protect the rights and welfare of human subjects involved in such investigations. |
| Part 312 | Investigational New Drug Application | This part contains procedures and requirements governing the use of investigational new drugs, including procedures and requirements for the submission to, and review by, the FDA of investigational new drug applications (IND's). |
| Part 314 | Regulations for Applications for FDA Approval to Market a New Drug (NDA Regulations) | This part sets forth procedures and requirements for the submission to, and the review by, the FDA of applications and abbreviated applications to market a new drug, as well as amendments, supplements, and post-marketing reports to them. |
*Resource: CFR regulations
Table 2: Some 21 CFR regulations covering GCP.
In order to provide a good practice of data monitoring, the data coordination and statistical analysis center is required and responsible for case report design, on-line data management system for data entry, editing and verification, and for the quality control of the conduct of the trial through training, certification, tracking of case report forms and reports, and design and maintenance of on-line analysis system for interim and final analyses. In practice, an independent data monitoring committee (DMC) is usually established for any confirmatory trials with planned interim analysis, in particular, for the trials conducted in life-threatening diseases or severely debilitating ailments [30]. It is usually adopted for the trials sponsored by government which consists of the disciplines in clinical, laboratory, epidemiology, biostatistics, data management and ethics. DMC are responsible to monitor the safety and ethical aspects of the trial with respect to the patients, investigators, sponsors, and the regulatory authorities in descending order of priority.
For the following paragraphs, we will point out few hot issues of recent development on clinical trials such as biomarker development for target clinical trial, translational research/medicine, and traditional Chinese medicine are also provided.
Personalized medicine
As it is well recognized that the U.S. Department of Energy (DOE) and the National Institutes of Health (NIH) had completed the Human Genome Project (HGP) for many years. Since the disease targets can be identified at the molecular level, it is foreseeable that the pathway to personalized medicine will utilize an individual’s full genomic sequence. The established molecular heterogeneity of human diseases requires the development of new paradigms for the design and analysis of randomized clinical trials as a reliable basis for predictive medicine [31]. Many top-tier medical institutions now have personalized medicine programs, and scientists and physicians are actively conducting clinical trials in genomic medicine. Since patients respond differently to medicines, and all medicines present the possibility of side effects. Based on the belief that these differences may be based on genetic factors, FDA has been providing scientific and strategic input to the International Serious Adverse Events Consortium (iSAEC) to identify genetic markers that are useful in predicting the risk of drugrelated serious adverse events. But the following regulatory science for evaluating the strategies and outcomes for personalized medicine are still need to be developed: (1) standards for whole genome sequencing; (2) fully qualified biomarkers (measurable characteristics in patients); and (3) innovative clinical trial designs and statistics.
Translational medicine/research
The NIH has been put into the effort to meet this challenge. One of the key is the transformation of translational clinical science which is closely combined with the novel interdisciplinary approaches, in order to promote human health. [32] With personalized medicine, pathological mechanisms can be investigated at the molecular genetic level. Translational research apply the findings on the molecular medicine from lab to the clinical research (from bench to bedside). In other words, it is a two-way research from the laboratory to patients which explore genes associated with the disease and its pathological mechanism to solve the problems about pharmaceutical development. Mankoff et al. [33] pointed out that there are three major obstacles to effective translational medicine in practice. The first is the challenge of translating basic science discoveries into clinical studies. The second hurdle is the translation of clinical studies into medical practice and health care policy. A third obstacle to effective translational medicine is philosophical Cosmatos and Chow [34]. It may be a mistake to think that basic science (without observations from the clinic and without epidemiological findings of possible associations between different diseases) will efficiently produce the novel therapies for human testing. Pilot studies such as non-human and non-clinical studies are often used to transition therapies developed using animal models to a clinical setting. Statistical process plays an important role in translational medicine. In this article, we follow Cosmatos and Chow [34] define a statistical process of translational medicine as a translational process for (1) determining association between some independent parameters observed in basic research discoveries and a dependent variable observed from clinical application, (2) establishing a predictive model between the independent parameters and the dependent response variable, and (3) validating the established predictive model. As an example, in animal studies, the independent variables may include in vitro assay results, pharmacological activities such as pharmacokinetics and pharmacodynamics, and dose toxicities and the dependent variable could be a clinical outcome (e.g., a safety parameter).
Traditional Chinese medicine
In recent years, the research on Chinese medicines (TCM) become the center of attention of many pharmaceutical companies, especially for those intended for treating critical and/or life-threatening diseases. A TCM is defined as a Chinese herbal medicine developed for treating patients with certain diseases as diagnosed by the four Chinese major techniques of inspection, auscultation and olfaction, interrogation, and pulse taking and palpation based on traditional Chinese medical theory of global dynamic balance among the functions/activities of all organs of the body [35]. Unlike evidence-based clinical research and development of a Western medicine (WM), clinical research and development of a TCM is usually experience-based with anticipated variability due to subjective evaluation of the disease under study Chow et al. [36]. The use of TCM in humans for treating various diseases has a history of more than five thousand years but no scientific documentation is available regarding clinical evidence of safety and efficacy of these TCMs.
In the past several decades, regulatory agencies of both China and Taiwan have debated which direction the TCM should take– Westernization or modernization [37,38]. Due to the fundamental differences between a WM and a TCM (Table 3), it is recognized that WMs tend to achieve the therapeutic effect sooner than that of TCMs for critical and/or life-threatening diseases. TCMs are found to be useful for patients with chronic diseases or non-life-threatening diseases. In many cases, TCMs have shown to be effective in reducing toxicities or improving safety profile for patients with critical and/ or life-threatening diseases. As a strategy for TCM research and development, it is suggested that (1) TCM be used in conjunction with a well-established WM as a supplement to improve its safety profile and/or enhance therapeutic effect whenever possible, and (2) TCM should be considered as the second line or third line treatment for patients who fail to respond to the available treatments. However, some sponsors are interested in focusing on the development of TCM as a dietary supplement due to (1) the lack or ambiguity of regulatory requirements, (2) the lack of understanding of the medical theory/mechanism of TCM, (3) the confidentiality of non-disclosure of the multiple components, and (4) the lack of understanding of pharmacological activities of the multiple components of TCM.
| Description | Western Medicine | Traditional Chinese Medicine |
|---|---|---|
| Active ingredient | Single | Multiple |
| Dose | Fixed | Flexible |
| Diagnostic procedure | Objective; validated | Subjective; not validated |
| Therapeutic index | Well-established | Not well-established |
| Medical mechanism | Specific organs | Global dynamic balance/harmony among organs |
| Medical perception | Evidence-based | Experience-based |
| Statistics | Population | Individual |
Table 3: Fundamental Differences between a WM and a TCM.
Since TCM consists of multiple components which may be manufactured from different sites or locations, the post-approval consistency in quality of the final product is both a challenge to the sponsor and a concern to the regulatory authority. As a result, some post-approval tests, such as tests for content uniformity, weight variation, and/or dissolution and (manufacturing) process validation, must be performed for quality assurance before the approved TCM can be released for use.
This research was partially supported by the Duke University Center for AIDS Research (CFAR), an NIH funded program (5P30 AI064518).