PMC/PubMed Indexed Articles

Indexed In
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- MIAR
- Euro Pub
- Google Scholar
Useful Links
Share This Page
Journal Flyer

Open Access Journals
- Agri and Aquaculture
- Biochemistry
- Bioinformatics & Systems Biology
- Business & Management
- Chemistry
- Clinical Sciences
- Engineering
- Food & Nutrition
- General Science
- Genetics & Molecular Biology
- Immunology & Microbiology
- Medical Sciences
- Neuroscience & Psychology
- Nursing & Health Care
- Pharmaceutical Sciences
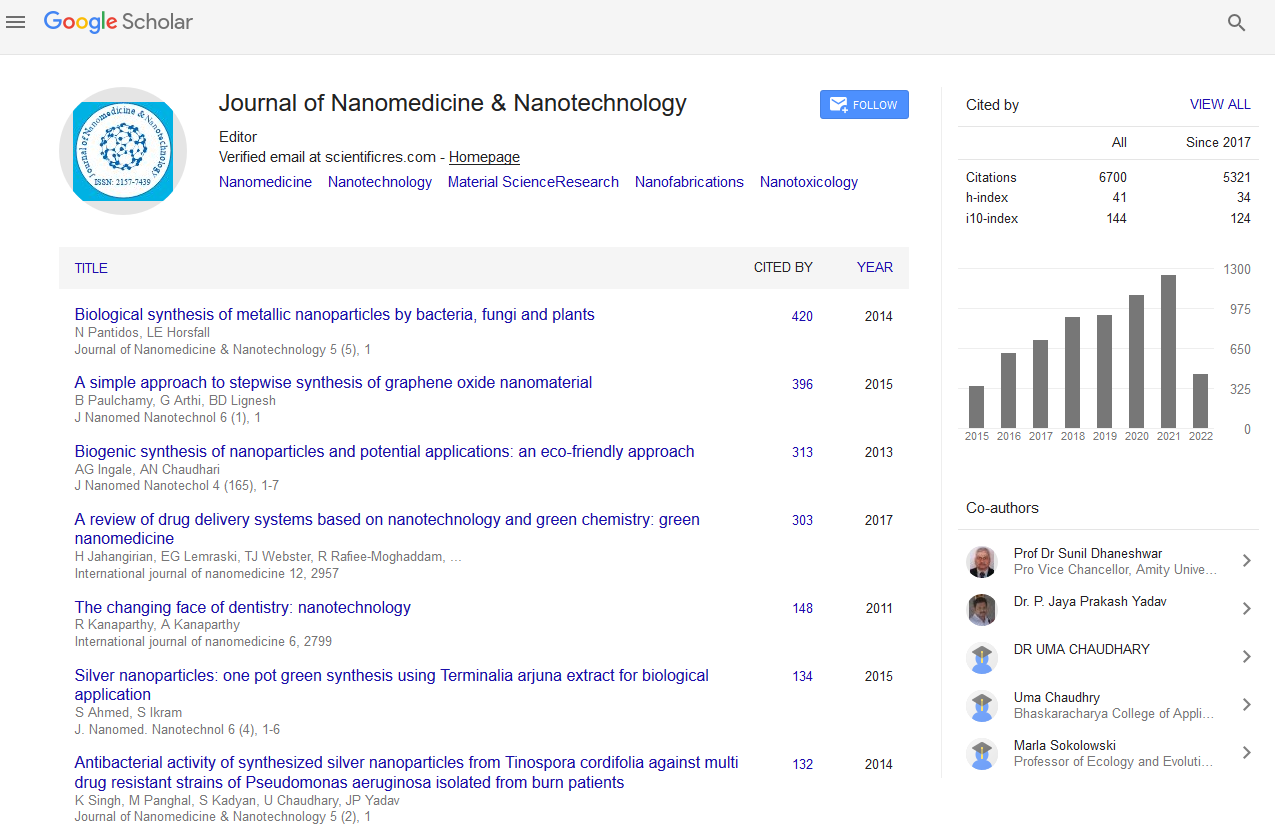
The structure and reactivity of potassium supported on SBA-15: Density functional theory study
World Congress on Petrochemistry and Chemical Engineering
November 18-20, 2013 Hilton San Antonio Airport, TX, USA
Daxi Wang, Bing Liu, Zhongxue Wang, Zhen Zhao and Jie Lan
Posters: J Pet Environ Biotechnol
Abstract:
The geometries, vibrational frequencies, electronic properties and reactivity of alkali metal supported on SBA-15 have been theoretically investigated by the density functional theory (DFT) method. The geometries of five kinds of alkali metals (Li, Na, K, Ru, Cs) supported on SBA-15 were optimized. The substitution energy was calculated in order to obtain the favored locations of the alkali atoms. For Li, Na and K, Alkali-O(2)-SBA-15 is the suitable model. While for Rb and Cs, Alkali-O(3)-SBA-15 is the suitable model. The vibrational frequencies of Si-O(1)-H and Si-O(3)-H on the K-O(2)-SBA-15 model are 2607 cm-1 and 3603 cm-1, which are in good agreement with those observed in experiment. Therefore, the model K-O(2)-SBA-15 is a reasonable model of the potassium supported on the SBA-15. The Mayer bond orders were calculated for Alkali/SBA-15. It is found that the Li-O(2), Na-O(2), K-O(2), Rb-O(3) and Cs-O(3) bonds possess the higher bond orders, which are in good accordance with the results of substitution energies and bond lengths. The K atom in K/SBA-15 shows strong positive electrostatic potential and the LUMO in K/SBA-15 is mainly contributed by the K atom, indicating that the K atom acts as the Lewis acid site in K/SBA-15. The oxygen molecule can be activated by on K/SBA-15 with an energy barrier of 103.19 kJ mol-1. It is found that the LUMO is mainly contributed by the activated oxygen atoms. The activated oxygen atoms become new Lewis acid center and are predicted to play an important role during catalytic reactions